Review
Development of Methodologies toward the Unified Synthesis of Ellagitannins
2024 Volume 72 Issue 4 Pages 349-359
Details
2024 Volume 72 Issue 4 Pages 349-359
Ellagitannins, a class of polyphenols with divergent structures, have attracted considerable attention from synthetic organic chemists. The basic structures in ellagitannins contain esters of D-glucose with galloyl or hexahydroxyldiphenoyl groups, as well as diaryl ether structures. Thus, the synthesis methodologies of such components have been developed by various groups, including our group. This review describes the synthetic methods reported by our group during 2017–2023, aimed at increasing the number of ellagitannins that can be chemically synthesized. In addition, recent related reports are introduced.
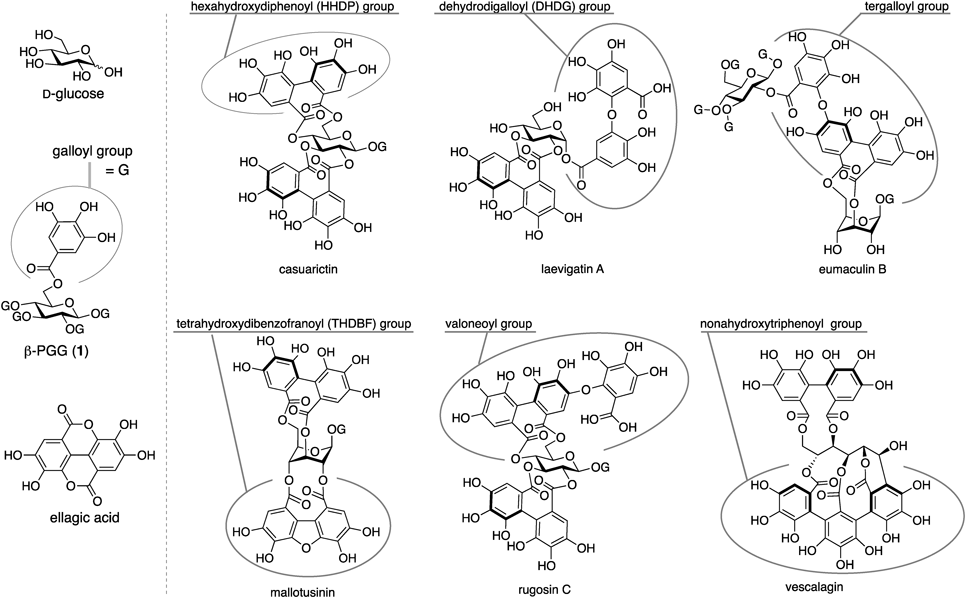
Ellagitannins are a class of polyphenols that have attracted attention in the fields of organic chemistry and biochemistry. This is because ellagitannins are structurally diverse, and a few of them exhibit remarkable biological activity.1–3) Their general structure components are D-glucose, galloyl and hexahydroxydiphenoyl (HHDP) groups (Fig. 1). These three motifs are connected through ester bonds because the HHDP groups are biosynthesized via a C–C coupling of two galloyl groups in pentagalloyl-D-glucose (β-PGG, 1).4–6) The origin name of ellagitannins is ellagic acid, which is a hydrolysis product of HHDP-bridged glucose. Importantly, approximately 40% of ellagitannins have a further critical component, namely C–O digallate structures. These structures contain a diaryl ether motif formed by a C–O bond connection between one galloyl group and another galloyl group or its analogue, such as an HHDP group.7) This major component can oligomerize monomeric ellagitannins, resulting in an extreme increase in the structural diversity of ellagitannins. In addition, other components, except for the aforementioned four, are biosynthesized, e.g., tetrahydroxydibenzofuranoyl (THDBF)8) and nonahydroxytriphenoyl groups.9) Owing to the presence of these components, more than >1000 ellagitannins have been isolated to date.
Prof. Yamada of Kwansei Gakuin University, who passed on in November 2019, had aimed to synthesize as many ellagitannins as possible, ideally all.10–13) The author joined Prof. Yamada’s group for 3 years from 2015. This review describes representative studies reported by Prof. Yamada and coworkers during 2017–2023 and briefly introduces recent reports by other groups.
The HHDP group in ellagitannins exhibits axial chirality due to the restricted rotation of the tetra-ortho-substituted aryl–aryl bond. The axial chirality tends to be controlled by the location of the HHDP bridge in D-glucose. For example, when the HHDP group is bridged between the oxygenated 3- and 6-positions (3-O and 6-O) of D-glucose, its axial chirality is generally R.2) Considering that the HHDP group is biosynthesized through β-PGG (1), the 3,6-O-(R)-HHDP-bridged glucose is biosynthesized via the flip of the pyranose ring of 1, as the conformation changes from an equatorial-rich chair to an axial-rich chair.
In 2017, we reported the biomimetic oxidative phenol coupling of 2, which is a partial benzyl (Bn)-protected compound of 1, and detected the 3,6-O-(R)-HHDP bridged glucose 3 in trace amounts14) (Chart 1a). This result implies that the 3,6-O-(R)-HHDP bridge could be synthesized via the oxidative phenol coupling of the 3,6-O-digalloyl glucose derivative 4. However, the reaction of 4 with CuCl2 and BuNH2 in MeOH did not afford the desired product, 5 (Chart 1b). This was attributed to the highly energetic barrier of flipping the pyranose ring from an equatorial-rich chair to an axial-rich chair. Thus, to construct a 3,6-O-(R)-HHDP bridge, we previously prepared acyclic digallate 6 via the opening of the pyranose ring of D-glucose and performed the CuCl2/BuNH2 oxidative phenol coupling reaction of 6.15) This reaction smoothly proceeded to produce the desired product 7 in 76% yield as the single isomer, which enabled the completion of the total synthesis of corilagin through the reconstruction of the pyranose ring (Chart 1c).

(a) Oxidative phenol coupling of partially Bn-protected compound of β-PGG. (b) Reaction of digallate 4 with a CuCl2/BuNH2 complex. (c) Previous synthesis of corilagin. PMB = p-methoxybenzyl.
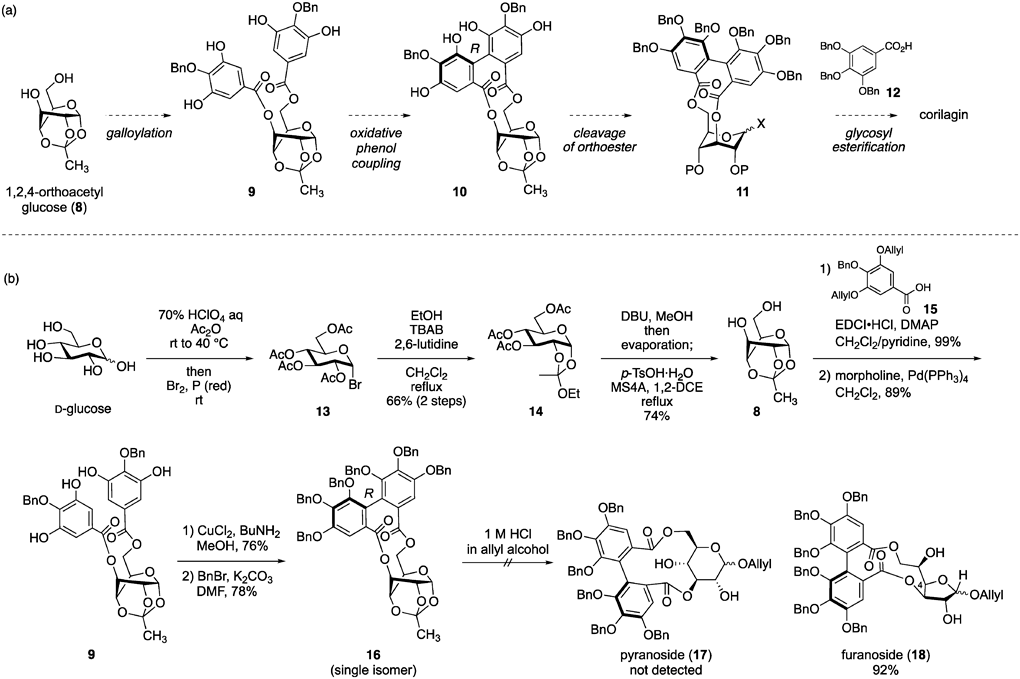
Because this method requires the opening and closing sequences of the pyranose ring, we modified the synthesis of the 3,6-O-(R)-HHDP-bridged glucose, using 1,2,4-orthoacetylglucose (8) (Chart 2a). This molecule is locked in an axial conformation by the orthoester bridge. Therefore, the two galloyl moieties in the 3,6-digalloylated compound 9 are proximal, which bodes well for the oxidative phenol coupling of 9. Furthermore, we suspected that the total synthesis of corilagin was enabled via the 1,2,4-orthoester cleavage of the coupling product 10, followed by the glycosyl esterification of 11 with a gallic acid derivative, 12.
(a) Modified synthetic plan of corilagin. (b) Oxidative phenol coupling of 9 and trial to cleave orthoester 16. EDCI·HCl = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, DMAP = 4-N,N-dimethylaminopyrdine, DMF = N,N-dimethylformamide, Ph = phenyl, Bu = butyl.
Although the synthesis of 8 is known,16,17) we established a synthesis method for producing >20 g of 818,19) (Chart 2b). After the full acetylation of D-glucose via treatment with a 70% HClO4 aqueous solution and acetic anhydride (Ac2O), Br2 and reddish phosphorus were added to the reaction mixture to generate PBr3 and HBr in situ, resulting in the α-selective bromination to produce 13.20) A subsequent reaction of 13 with tetrabutylammonium bromide (TBAB), EtOH, and 2,6-lutidine in a refluxed CH2Cl2 solution induced an intramolecular attack on the anomeric position by the carbonyl oxygen in the 2-O acetyl (Ac) group. This was followed by the capture of the generating oxocarbenium ion with EtOH, affording 14 in 66% yield over two steps. The three Ac groups in 14 were removed using 1,8-diaza[5.4.0]-7-undecene (DBU) and MeOH. The resulting crude product in 1,2-dichloroethane (1,2-DCE) was treated with p-toluenesulfonic acid monohydrate (TsOH·H2O) in the presence of molecular sieve 4A (MS4A), producing 8 in 74% yield via intramolecular orthoester formation.
Following our synthetic strategy, we constructed the 3,6-O-(R)-HHDP bridge using 821) (Chart 2b). Two galloyl moieties were incorporated into 8 under typical condensation reaction conditions using carboxylic acid 15, and the subsequent Pd(0)-mediated deallylation afforded 9 in high yield. Favorably, the oxidative phenol coupling of 9 proceeded smoothly with CuCl2 and BuNH2 in MeOH, and the resulting compound was fully benzylated to provide the 3,6-O-(R)-HHDP-bridged glucose 16 as a single isomer. This result appeared to be a critical breakthrough in constructing the 3,6-O-(R)-HHDP-bridged glucose; however, the next orthoester cleavage was problematic. Thus, 16 was subjected to acidic alcoholysis to afford furanoside 18 in 92% yield, not pyranoside 17. The production of 18 appeared to occur via the intramolecular attack on the generating anomeric oxocarbenium ion by releasing the 4-O hydroxy group, followed by the opening of the resulting acetal moiety. We found certain reaction conditions to produce pyranoside selectively; however, the methods were inefficient.
Fortunately, this problem was resolved in another research project—the synthesis of thioglucoside 19 bearing the 3,6-O-[1,1′-(ethane-1,2-diyl)dibenzene-2,2′-bis(methylene)] (EDB) bridge.22,23) We previously reported the orthoether cleavage of 1,2,4-O-orthoacetyl-3,6-O-(o-xylylene)glucopyranoside using PhSH and trimethylsilyl triflate (TMSOTf) at −40 °C.24) Adoption of the reaction conditions to 3,6-O-EDB-bridged-glucose 20, followed by methanolysis of the resulting Ac group afforded the desired compound 19 (entry 1 in Table in Chart 3a). However, this method produced an α/β = 32/68 anomeric mixture and suffered from the reproducibility for the yield of 19. To improve this result, we screened the activator and revealed that the use of BF3·OEt2 afforded a result similar to that of TMSOTf (entry 2). Contrarily, the selection of InBr3 enabled a dramatical increase in the yield of 19 with high β-selectivity, even with a decrease in the amount of PhSH (entry 3). Furthermore, the use of benzotrifluoride as a solvent enabled the cleavage reaction of the orthoester at room temperature and increased the yield of 19 to 96% (entry 4). Pleasingly, the adoption of this reaction system for orthoester 16 also afforded acetate 21 in 97% yield as a single isomer21) (Chart 3b).

(a) Orthoester cleavage of 20 with a 3,6-O-EDB bridge. (b) Transformation of 16 into 21.
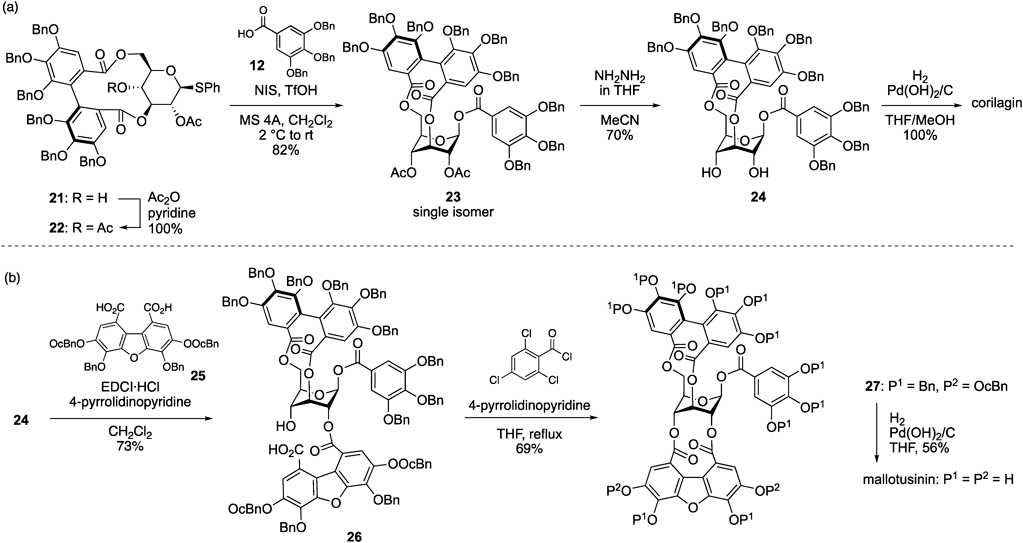
The conversion of 21 into corilagin was achieved in four steps (Chart 4a). Compound 22, an acetylation product of 21, was subjected to glycosyl esterification with tri-O-benzylgallic acid (12) using N-iodosuccinimide (NIS) and triflic acid (TfOH)25,26) to provide a β-galloyl ester, 23, in 82% yield as a single isomer. The chemoselective removal of the two Ac groups of 23 in the presence of the galloyl and HHDP ester occurred via hydrazinolysis, and hydrogenolysis of the resulting diol 24 completed the total synthesis of corilagin.21)
(a) Modified synthesis of corilagin. (b) Total synthesis of mallotusinin. THF = tetrahydrofuran.
Next, we synthesized other ellagitannins, in which the 2-O and 4-O on corilagin are bridged on another component because such ellagitannins have been isolated in nature27–32) but have never been chemically synthesized. The target natural product was set as mallotusinin with a THDBF group.8) Its total synthesis was achieved via a two-step bislactonization between diol 24 and dicarboxylic acid 25, which bears a dibenzofuran framework21) (Chart 4b). The two p-octylbenzyl (OcBn) groups33) in 25 were incorporated to enhance the solubility of 25 in an organic solvent. Mono-acylation proceeded smoothly using EDCI·HCl and 4-pyrrolidinopyridine34) to provide seco acid 26 chemoselectively. In this reaction, the choice of 4-pyrrolidinopyridine as an additive was a critical factor in producing 26 with a satisfactory yield. The intramolecular lactonization of 26 occurred efficiently upon treatment with the Yamaguchi reagent35) and 4-pyrrolidinopyridine at reflux, affording 27. The subsequent removal of the Bn and OcBn groups in 27 by hydrogenolysis completed the total synthesis of mallotusinin.
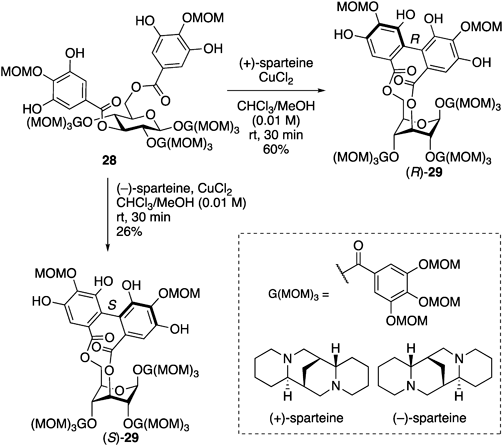
After the publication of this work, the Kawabata group reported a remarkable strategy for constructing the 3,6-O-HHDP-bridged glucose36) (Chart 5). They employed an oxidative phenol coupling of digallate 28, which exhibits a similar structure similar to that of 2, using CuCl2 and a chiral amine, (+)-sparteine. The reaction conditions enabled the flip of the pyranose ring, and the subsequent oxidative phenol coupling proceeded swiftly to afford (R)-29 in 60% yield as a single isomer. Furthermore, the change of (+)-sparteine to (−)-sparteine resulted in the production of its diastereomer, (S)-29, in 26% yield. These results indicated that the Kawabata method can stereodivergently afford (R)-29 and (S)-29.
Work of Kawabata group. MOM = methoxymethyl.
As mentioned in section 2, the axial chirality of the HHDP group tends to be controlled by its location on glucose. For the 4,6-O-HHDP bridge in glucose, its axial chirality is extremely S, although more than 300 ellagitannins with 4,6-O-HHDP bridges have been isolated in nature.2) This bias tendency was also reflected in the chemical synthesis of the 4,6-O-HHDP-bridged glucose37–49) (Chart 6). For examples, the oxidative phenol coupling of 4,6-digallate 30 under typical reaction conditions with CuCl2 and BuNH2 in MeOH predominantly occurred with (S)-selectivity to afford (S)-31 in quantitative yield.42) Recently, the Wakamori group reported that the oxidative phenol coupling of 30 using another copper-amine complex, [Cu(OH)TMEDA2]Cl2,50,51) in CH2Cl2 kinetically produced (R)-31.52) However, its isomerization into (S)-31 gradually occurred over time and finally isolated (S)-31 in 85% yield after 90 min. As another approach for constructing the 4,6-O-HHDP-bridged glucose, the double esterification of 4,6-diol, such as 32, with HHDP-derived carboxylic acid has been reported.14,53–57) However, the reaction using 32 and rac-33 only produced (S)-34, and (R)-34 was not detected because the intramolecular lactonization of seco acid derived from 32 and (R)-33 was disfavored because of the bridge strain.58,59)

(a) Oxidative phenol coupling of 4,6-digallate 30. (b) Reported double esterification of 32 with rac-33. TMEDA = N,N,N′,N′-tetramethylethylenediamine.
These results indicated that the efficient chemical synthesis of the 4,6-O-(R)-HHDP-bridged glucose is a challenging issue. In addition, the presence of four natural ellagitannins with a 4,6-O-(R)-HHDP group,60–62) one of which is neostrictinin,62) inspired us to tackle the formation of the disfavored bridge via a chemical approach. To obtain the irregular HHDP bridge, we employed a two-step bislactonization strategy63) via the mono-acylation of diol 35 with acid anhydride (R)-36,64) followed by intramolecular lactonization of seco acid 37. Product (R)-38 will easily lead to the formation of neostrictinin via the introduction of a galloyl group at the anomeric position (Chart 7).
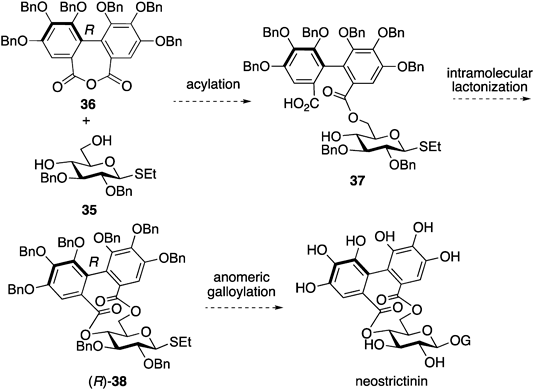
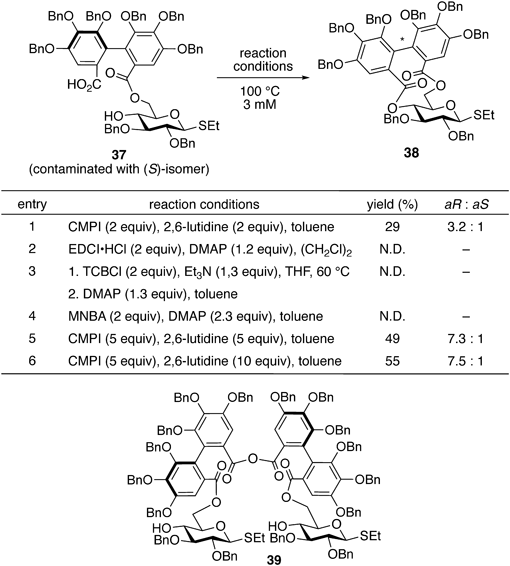
The screening of the reaction conditions for the intramolecular lactonization of 37 revealed that the use of 2-chloro-N-methylpyridinium iodide (CMPI)65) and 2,6-lutidine in diluted toluene afforded the desired product in low yield66) (entry 1 in Chart 8). This screening used 37 prepared from 35 and 87% enantiomeric excess (ee) of 36; therefore, the product contained (R)-38 and (S)-38 as a 3.2 : 1 diastereomeric ratio. Other typical condensation reagents, such as EDCI·HCl; 2,4,6-trichrolobenzoyl chloride (TCBCl)35); and 2-methyl-6-nitrobenzoic anhydride (MNBA),67) did not produce 38 (entries 2–4). An increase in the amount of CMPI and 2,6-lutidine improved the yield of 38 (entry 5). Finally, the use of 5 and 10 equivalent (equiv.) of CMPI and 2,6-lutidine at 100 °C, respectively, afforded the best result of 38 in 55% isolated yield in a 7.5 : 1 diastereomeric ratio (entry 6). The monitoring of this reaction via NMR spectroscopy indicated that the CMPI-mediated intramolecular lactonization followed an unusual reaction pathway because dimeric acid anhydride 39 was first generated. Afterward, the transformation of 39 into 38 was observed. The exact reaction mechanism of the intramolecular lactonization reaction remains ambiguous.
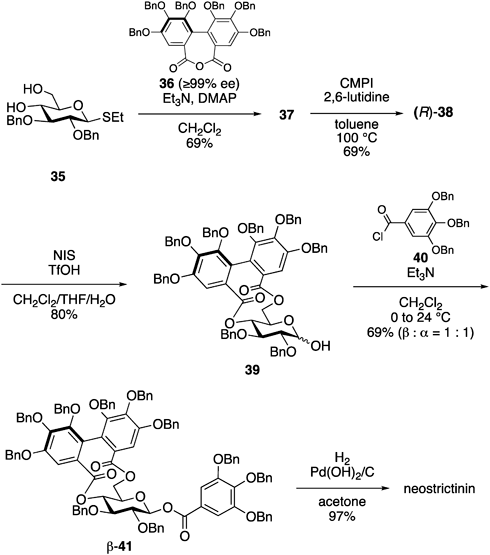
Based on the established method for constructing the 4,6-O-(R)-HHDP structure, the total synthesis of neostrictinin was performed using enantiomerically pure 3666) (Chart 9). After preparing seco acid 37 using (R)-36 and diol 35, the treatment of 37 under optimized reaction conditions (entry 6 in Chart 8) produced (R)-38 as a single isomer in 69% yield. The hydrolysis of the anomeric ethylthio group using NIS and TfOH in the presence of water afforded hemiacetal 39. The subsequent acylation of the 1-hyrdoxy group of 39 with tri-O-benzyl galloyl chloride (40) afforded a 1 : 1 anomeric mixture of 41 in 69% yield. After separating the anomeric mixture by silica gel chromatography, β-41 was subjected to hydrogenolysis, yielding neostrictinin.
C–O digallate structures are components, including diaryl ether moieties, which are formed over two galloyl groups or their derived groups.57) Therefore, various components are found in ellagitannins2,7) (Fig. 2a). The dehydrodigalloyl (DHDG) group has a simple component because it comprises the two galloyl groups. An ethereal linkage is formed between the 3-O hydroxy group in one and the C-2 carbon in the other. The iso-DHDG group, where the location of the ether bond differs from the DHDG group, has been found in nature. The diaryl ether structures formed among the galloyl and HHDP groups are more diverse because connecting patterns between the HHDP group and galloyl groups are more abundant than those between the two galloyl groups. The valoneoyl, tergalloyl, and macaranoyl groups are representative components, each of which bears an ether bond corresponding to the 4-O, 5-O, and 6-O positions of the HHDP group, respectively. More complicated components exist, where more than two C–O bonds are installed in C–O digallate structures, such as the hellinoyl group.68) Carboxyl groups corresponding to galloyl groups in the C–O digallate structures can function during the oligomerization of a monomeric ellagitannin via esterification with a hydroxy group of a glucose moiety in other ellagitannins. A few of such structures are shown in Fig. 2b. The broad structural diversity of the ellagitannins originates from C–O digallate structures, suggesting that a unified synthesis strategy of these components is essential for aiming at the synthesis of various ellagitannins.

Among reports on the synthesis of C–O digallate structures,40,45,47,48,69–76) our method reported enables the synthesis of various structures.57,74,75) In 2014, Prof. Yamada reported a unified synthesis methodology, which can furnish the DHDG, valoneoyl, and tergallloyl groups, using the oxidative compound of a gallic acid derivative57) (Chart 10). Thus, galloyl ester 42 was transformed into the corresponding brominated aldehyde 43, which was subjected to oxidative dearomatization77) using phenyliodine(III) bis(trifluoroacetate) (PIFA) and benzyl alcohol (BnOH) to provide ortho-quinone monoketal (o-Qk), 44. This product reacted with a phenolate ion via the oxa-Michael addition and the simultaneous elimination of the bromide ion to form the C–O bond. The resulting coupling product, 45, induced reductive aromatization using Pd(0) and Et3SiH to afford phenol 46. o-Qk 47 bearing an ester moiety did not permit the oxa-Michael addition from the phenolate ion, indicating that the aldehyde moiety in 44 was essential for the formation of the C–O bond. The presence of the aldehyde moiety was advantageous because its selective transformation toward the connection to other monomeric ellagitannins can be employed in the presence of other ester moieties in diaryl ether components. However, regarded as the redox economy,78) the method for transforming 42 into 46 requires improvement.
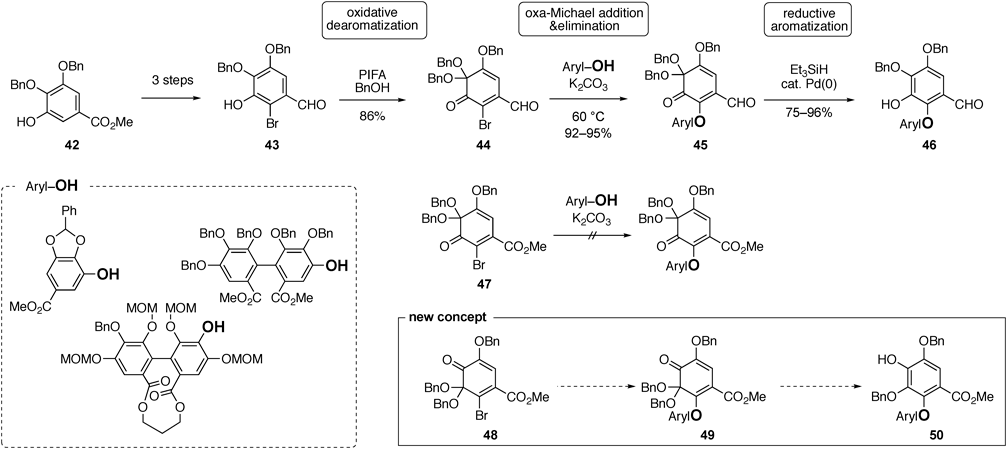
Considering this disadvantage, we created a novel o-Qk with higher electrophilicity than ester 47.74) Although 47 has two electron-withdrawing groups, an ester and a ketone, the latter function appears to not involve a decrease in the electrophilicity of 47 because it is at a neighboring position from the bromine-substituted carbon. Thus, we designed o-Qk 48, in which the ketone and ketal locations in 47 were reversed. This change enhanced the electrophilicity of the bromine-substituted carbon by acquiring an additional conjugation effect from the ketone, which allowed the oxa-Michael/elimination reaction of phenolate ions. The reductive aromatization of the coupling product, 49, would smoothly occur similar to that of 45, producing phenol 50.
Chart 11 shows the synthetic route of 48.74) The chemoselective allylation of methyl gallate (51) using KHCO3 as a base in the presence of catalytic KI42) afforded diol 52. The bromination of 52 by 1,3-dibromo-5,5-dimethylhydantoin produced a mixture of desired product 53 and overreacted product 54, along with the recovery of 52. After the mixture was benzylated, the two products were separated via silica gel chromatography, and 55 and 56 were furnished in 48 and 41% yields, respectively, over two steps. The Pd-mediated deallylation of 55, followed by the oxidative dearomatization of 57 using PIFA in the presence of BnOH, was employed. The oxidation reaction generated two oxocarbenium ion intermediates, 58 and 58′, both of which reacted with BnOH, to produce 48 and its regioisomer, 48′, in 54 and 40% yields, respectively. Fortunately, the desired o-Qk 48 was stable in air at ambient temperature; however, 48′ decomposed at the temperature.
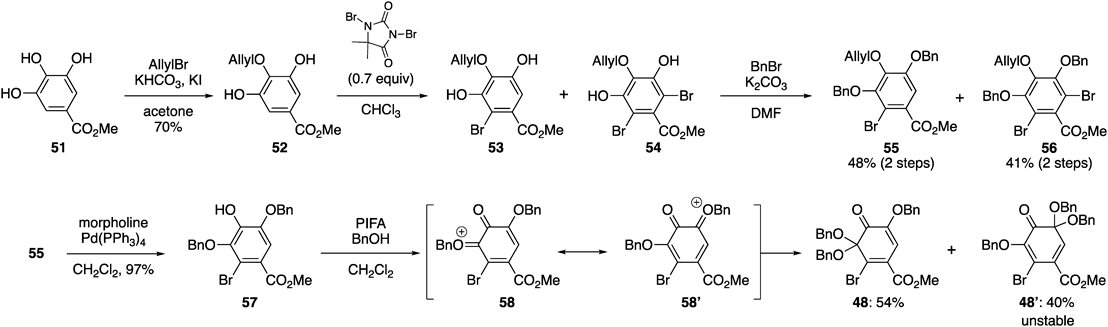
The oxa-Michael addition of various phenolate ions to 48 and the elimination of the bromide ion proceeded smoothly74) (Chart 12). For the DHDG group, phenol 59 was used as the nucleophile, and the reaction of 59 with 1.3 equiv. of 48 and K2CO3 in acetonitrile occurred at 70 °C to afford 60 in 95% yield. Although the reductive aromatization of 60 occurred under the reaction conditions for transforming 45 into 46, we revealed that a sodium borohydride reduction, followed by treatment with hydrochloric acid, produced 61 in excellent yield. In addition, the hydrogenolysis of 60 using the Pearlman catalyst was applicable for the reductive aromatization, which simultaneously removed the Bn groups to furnish pentaphenol 62. When using the HHDP-derived phenol, 63, for the oxa-Michael addition reaction with 48, the desired C–O bond was efficiently formed using 3.0 equiv. of 48. The resulting compound, 64, was subjected to hydrogenolysis to afford 65 with a valoneoyl group. During the construction of the tergalloyl group, the use of 48 afforded a better result than that of 44. Thus, the previous method using 44 did not arise in the reaction of 66; therefore, we used the less sterically hindered phenol, where three Bn groups of 66 was replaced with MOM groups (the structure is shown in Chart 10).57) Furthermore, the reaction solvent was changed from MeCN into DMSO to enhance the reactivity of the phenolate ion. Oppositely, the reaction of 66 with 48 proceeded smoothly in MeCN to furnish 67 in 84% yield, indicating a higher reactivity of 48 compared with that of 44. The hydrogenolysis and subsequent methylation of the resulting phenols upon treatment with diazomethane afforded the tergalloyl structure, 68.
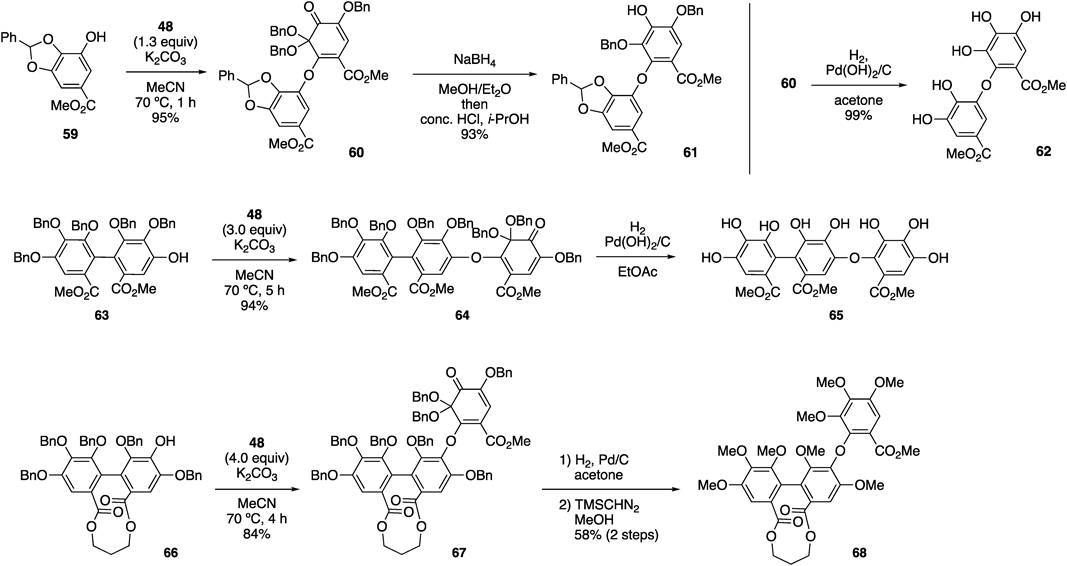
Remarkably, the novel method was applicable to form the macaranoyl structure, which cannot be synthesized using the previous method75) (Chart 13a). Thus, 48 reacted with phenol 69 (method A), the hydroxy group of which is at the most crowded position of the HHDP group, affording the coupling product, 70. However, because the reaction occurred over five days, byproducts derived from 48 were detected under the reaction conditions, resulting in the decrease in the yield of 70. The byproducts included 71, and its production occurred because of the oxa-Michael addition/elimination reaction of 48 and phenol 72, which was generated via the self-aromatization of 48 under light irradiation (Chart 13b). We hypothesized that this side reaction would be suppressed in the dark, and that the addition of 18-crown-6 would enhance the reactivity of the phenolate ion derived from 69, which increased the yield of 70. Fortunately, the reaction was completed within 49 h (method B), affording 70 in 66% yield. The subsequent reductive aromatization of 70 proceeded smoothly under Luche reduction conditions to furnish phenol 73. We successfully synthesized 74 with a macaranoyl structure using o-Qk 75 and phenol 76, both of which were more sterically hindered than 48 and 69, through the oxa-Michael elimination reaction under reaction conditions similar to those of method B. The resulting product underwent the synchronously reductive aromatization of the o-Qk moiety and the removal of all the Bn groups via hydrogenolysis (Chart 13c).
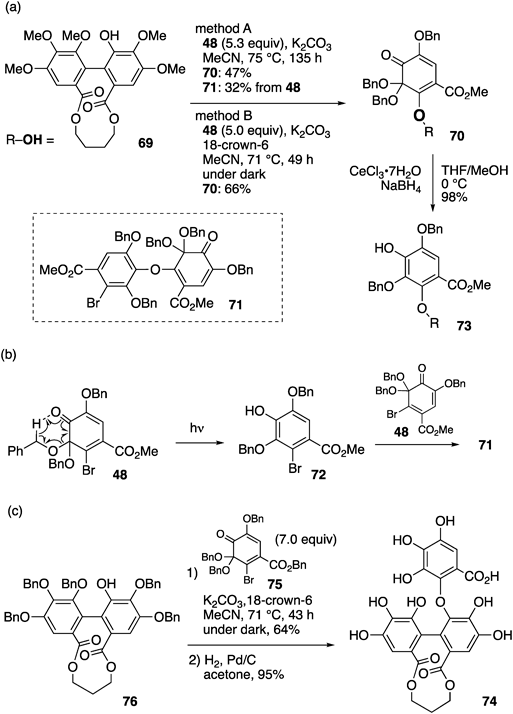
(a) Construction of the macaranoyl structure. (b) Fluorescent-light induced self-aromatization of o-Qk 48. (c) Application in the synthesis of 74.
We applied the C–O digallate structure synthesis to the total synthesis of rugosin C49) (Chart 14a). The Michael addition of phenol 77 to o-Qk 75, followed by the elimination of the bromine atom, produced 78 in high yield. Hydrogenolytic conditions induced the reductive aromatization of the o-Qk moiety in 78 and the removal of all the Bn groups to afford rugosin C in 73% yield. The Wakamori group recently achieved the synthesis of macaranin B using similar synthesis strategy79) (Chart 14b). The reaction of 79 with 75, the generating compound of which was subjected to the reductive aromatization conditions using sodium borohydride followed by acidic hydrolysis and benzylation, providing 80 in 66% over three steps. The subsequent four steps including anomeric galloylation, and removal of all Bn groups completed in the total synthesis of macaranin B.
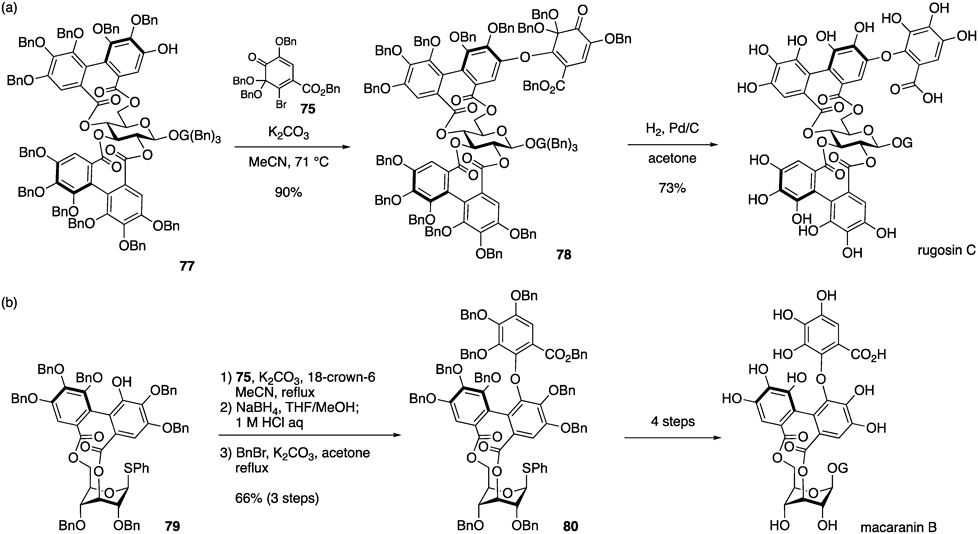
(a) Our report for rugosin C. (b) Wakamori report for macaranin B.
This paper describes the novel methods toward the unified total synthesis of ellagitannins. The efficient synthesis method of the 3,6-(R)-O-HHDP bridge is expected to develop the total synthesis of ellagitannins with an additional bridge between the 2-O and 4-O hydroxy group, except for mallotusinin. Although the 4,6-(R)-O-HHDP bridge is a rare component in ellagitannins, our result might be applicable in the synthesis of natural products with a macrocycle, the formation of which is disfavored. Our modified synthesis of C–O digallate structures can be employed in the formation of various diaryl ether structures and total syntheses of ellagitannins containing such components. It is expected that our methods will contribute to the development of organic synthetic chemistry of ellagitannins.
I would like to express my sincere gratitude to Prof. Dr. Hidetoshi Yamada, who suddenly passed away on November 23, at Kwansei Gakuin University. Among the periods where I joined his group, I received significant guidance and encouragement from him in relation to this review, resulting in production of numerous works for ellagitannin chemistry. I would like to devote myself based on what I learned from him. In addition, I deeply appreciate all the students, who were involved in this study. I also thank to Dr. Wakamori (Tokyo University of Agriculture) and Prof. Tanino (Hokkaido University), for a lot of considerable supports in this study to me. This research was funded by JSPS KAKENHI, Grant numbers JP16KT0061, JP19K15549, and JP16H01163 in Middle Molecular Strategy, and the MEXT-supported the program for the Strategic Research Foundation at Private Universities (S1311046).
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2023 Pharmaceutical Society of Japan Award for Young Scientists.