Abstract
Chagas disease, a neglected tropical disease caused by the protozoan Trypanosoma cruzi poses a significant health challenge in rural areas of Latin America. The current pharmacological options exhibit notable side effects, demand prolonged administration, and display limited efficacy. Consequently, there is an urgent need to develop drugs that are safe and clinically effective. Previously, we identified a quinone compound (designated as compound 2) with potent antiprotozoal activity, based on the chemical structure of komaroviquinone, a natural product renowned for its antitrypanosomal effects. However, compound 2 was demonstrated considerably unstable to light. In this study, we elucidated the structure of the light-induced degradation products of compound 2 and probed the correlation between the quinone ring’s substituents and its susceptibility to light. Our findings led to the discovery of quinones with significantly enhanced light stability, some of which exhibiting antitrypanosomal activity. The most promising compound was evaluated for drug efficacy in a mouse model of Chagas disease, revealing where a notable reduction in blood parasitemia.
Introduction
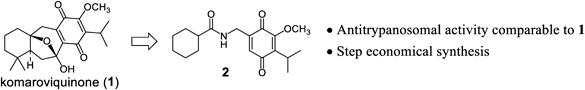
Chagas disease, caused by the protozoan parasite Trypanosoma cruzi (T. cruzi), is endemic in Latin America and has seen an increasing global prevalence due to the migration of chronically infected individuals.1) WHO estimates a worldwide infection rate of approximately 6–7 million people, leading to an annual average of 10000 related deaths.2) The disease manifests in both acute and chronic stage, with most infected adults experiencing mild symptoms during the acute stage, followed by the progression to the chronic stage. It is estimated that around 30% of patients develop symptomatic diseases, often associated with serious health conditions such as cardiomyopathy and gastrointestinal disease.3) The current therapeutic arsenal for Chagas disease comprises two nitroaromatic heterocycles, namely nifurtimox and benznidazole. While both drugs demonstrate efficacy during the acute phase of the disease, they are associated with severe side effects,4) and their effectiveness against the chronic form is limited.5) Recently clinical trials assessed the potential of two azole drugs, posaconazole6,7) and fosravuconazole (E1224),8) known for inhibiting the enzyme CYP51 involved in ergosterol biosynthesis. Despite a significant reduction in parasitemia during the treatment period, the prevalence of relapses was notable, raising concerns about the suitability of drug class for future development. Therefore, there is an urgent need for the development of safe and effective antiparasitic drugs.9) In pursuit of a new therapeutic agent for the Chagas disease, we turned our attention to the natural product komaroviquinone (1),10,11) renowned for its antitrypanosomal activity. Specifically, our investigation led to the identification of a quinone compound (compound 2, Fig. 1) that could be synthesized through a shorter synthetic route than (1), yet exhibited comparable antiprotozoan activity.12,13) In this study, we describe the characterization of quinone compounds structurally related to compound 2 aiming to refine and enhance its properties. All quinone derivatives were examined for their antitrypanosomal activity against four different T. cruzi strains, i.e., Sylvio, Tulahuen, Esmeraldo, and CL Brener. Additionally, we conducted in vivo studies employing a murine model infected with the Y strain.
Results and Discussion
Compound 2 exhibited antitrypanosomal activity comparable to that of compound 1, however, like compound 1, it displayed light-sensitive and thus unstable.14) When subjecting a 100 µM aqueous solution of compound 2 to close range (approximately 8 cm) fluorescent light irradiation at room temperature, over 60% of compound 2 degraded within 1 h (Table 1). Recognizing the light instability of a compound during evaluation necessitates careful consideration of light exposure when assessing various properties. Additionally, supplementary investigations, such as assessing the toxicity of decomposition by-products, may become imperative. To enhance light stability, the initial step involved identifying the chemical structure of the degradants to pinpoint the susceptible moiety of compound 2 to light-induced degradation.
Table 1. Decomposition of Compound
2 by Light Irradiation
a) Remaining % was calculated as the peak area ratio at 15, 30, and 60 to 0 min.
To elucidate the impact of fluorescent light on compound 2, an aqueous solution was exposure to irradiation, and the primary degradation products were analyzed using LC-MS to discern their chemical structures. The molecular weight of a degradation product was found to be 14 units smaller than that of compound 2, and its retention time was shorter, implying a more hydrophilic nature. Additionally, the decomposed product was isolated by column chromatography, and 1H-NMR measurements unveiled the disappearance of the singlet peak at 3.94 ppm, attributable to the methoxy group of compound 2. Collectively, these findings indicate that light irradiation induced the demethylation of the methoxy group in compound 2, leading to the formation of compound 3 (Fig. 2).
Subsequently, an investigation was conducted to explore the relationship between various substituents on the quinone ring and their stability to light,15) while preserving the essential quinone structure for antitrypanosomal activity.13) The photostability of compounds 4–7, lacking methoxy groups was evaluated, revealing that these compounds decomposed at a notably slower rate and displayed higher photostability compared to compound 2 (Table 2). Similarly, the light stability of compounds 8–10, bearing methoxy groups, was evaluated, demonstrating minimal degradation. The unexpected observation that compounds 8–10, with methoxy groups similar to compound 2, exhibited minimal decomposition under fluorescent light is not fully understood, and the underlying mechanism remains unclear. Nevertheless, addressing the issue of light instability could be achieved by ensuring that the substituents on the quinone ring adopted the same substitution mode as observed in compounds 8–10.
Table 2. Decomposition of Quinone Compounds
4–
10 upon Light Irradiation
a) Remaining % was calculated as the peak area ratio at 15, 30, and 60 to 0 min.
Next, compounds 5–10 were evaluated for their antitrypanosomal activity, cytotoxicity, and metabolic stability utilizing liver microsomes (Table 3). Compound 5, featuring two methyl groups on the quinone ring, exhibited slightly higher activity than compound 2 against all four tested strains. Conversely, compound 6, an isomer of compound 5, displayed slightly lower activity than compound 2, except in the case of the CL Brener strain. Compound 7, bearing three methyl groups on the quinone ring, demonstrated significant antiprotozoal activity against Esmeraldo, but exhibited lower activity against Sylvio and Tulahuen, ultimately displaying weaker overall antiprotozoal activity compared to compound 2. The stability of compounds 5–7 in the presence of liver microsomes was comparable. Compound 8 showed little or no antiprotozoal activity against the different strains, except for Tulahuen. However, compound 8 displayed extreme instability in metabolic reactions with liver microsomes, hindering the determination of its metabolic rate. This suggests a high susceptible to intracellular degradation, potentially explaining its lack of antiprotozoal activity against T. cruzi. Compound 9, an isomer of compound 8, exhibited reasonable stability in the presence of liver microsomes but showed lower antiprotozoal activity than compound 2 against all four tested strains. Compound 10, featuring an additional methyl group compared to compound 8, demonstrated satisfactory metabolic stability and slightly lower antiprotozoal activity against Tulahuen strain than compound 2, but similar activity against Sylvio, and robust antiprotozoal activity against Esmeraldo and CL Brener. Importantly, none of the compounds showed significant cytotoxicity to mammalian cells, with compounds 6, 9, and 10 particularly showing no cytotoxicity, at least at the highest concentration tested at 100 µM.
Table 3. Antitrypanosomal Activity, Cytotoxicity to Mammalian Cells, and Metabolic Stability of Quinone Compounds
| Compound | IC50 (µM) a) | HT1080CC50 (µM) | Cli (mg/min/kg)b,c) |
|---|
| Sylvio | Tulahuen | Esmeraldo | CL Brener | Human | Mouse |
|---|
| 2 | 1.7 | 0.50 | 4.0 | 2.5 | N.D. | N.D. | N.D. |
| 5 | 1.0 | 0.17 | 1.0 | 0.80 | 45 | 497 | 2084 |
| 6 | 3.8 | 1.3 | 7.7 | 1.8 | >100 | 288 | 2490 |
| 7 | 14.6 | 27.4 | 0.50 | 2.2 | 51 | 286 | 3002 |
| 8 | >100 | 18.7 | >100 | >100 | 46 | N.C. | N.C. |
| 9 | 35.8 | 79.7 | 18.5 | 16.9 | >100 | 259 | 1013 |
| 10 | 1.9 | 1.9 | 0.10 | 0.10 | >100 | 421 | 1830 |
| BZLd) | 1.9 | 2.2 | 1.4 | 6.6 | N.D. | N.D. | N.D. |
a) The IC50 values were calculated by nonlinear regression using GraphPad Prism version 9.0. These data represent the averaged of data normalized with wells containing dimethyl sulfoxide (DMSO) (0% inhibition) and 100 µM benznidazole (100% inhibition) as the negative and positive controls, each measured in quadruplicates. b) Hepatic microsomal stability was assessed for each compound in duplicate. c) N.D.: Not determined. N.C.: Not calculable because the compound degraded during the 5-min pre-incubation. d) BZL: benznidazole.
Based on these findings, we decided to further conduct in vivo studies using a mouse model of Chagas disease using compound 10. This compound exhibited a high antiprotozoal activity and satisfactory metabolic stability, with minimal degradation due to light exposure. C57BL/6 mice were intraperitoneally injected with the Y strain trypomastigotes, followed by intraperitoneal injections of either benznidazole or compound 10 twice a week, starting from the day after infection. T. cruzi is no longer observed in the blood a few days after infection and is observed again in the blood after about 2 weeks. Therefore, we first measured blood parasitemia on day 16 post infection, but no protozoa was observed in the blood at that stage. Later, on day 18, we measured parasitemia, and found that some mice in the control group had high levels of parasitemia, and further, on day 19, very high levels of protozoa were observed (Fig. 3). On day 20 post infection, the mice in the control group that showed high parasitemia died, so the subsequent parasitemia are not shown in the graph. In contrast to the control group with elevated parasitemia, both the benznidazole- and compound 10-treated groups displayed lower parasitemia and all mice survived throughout the measurement period.
Conclusion
In this study, we identified a new compound (10), which demonstrated remarkable photostability alongside with excellent in vitro antitrypanosomal activity. The effectiveness of compound 10 was further assessed in a murine model of Chagas disease, revealing therapeutic effects on par with the approved antiparasitic drug benznidazol. Our future efforts will focus on exploiting 10 as a lead compound to undertake a systematic structure–activity relationship, with the ultimate goal of developing analogues with even greater therapeutic efficacy.
Acknowledgments
This research was partially supported by a Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under the Grant Number JP23ama121053. Grants-in-Aid for Scientific Research (A) No. 20H00620, and (B) No. 23H02711 to D.K.I. from the Creative Scientific Research Grant from the Japan Society for the Promotion of Science (JSPS); and a Grant from The Leading Initiative for Excellent Young Researchers (LEADER) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) (No. 16811362 to D.K.I.). We thank the staff at Bioresource Center, Gunma University Graduate School of Medicine for technical help.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
References and Notes
- 1) Lidani K. C. F., Andrade F. A., Bavia L., Damasceno F. S., Beltrame M. H., Messias-Reason I. J., Sandri T. L., Front. Public Health, 7, 166 (2019).
- 2) “WHO Chagas disease (American trypanosomiasis).”: ‹https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis)›, cited 20 December, 2023.
- 3) Pérez-Molina J. A., Norman F., López-Vélez R., Curr. Infect. Dis. Rep., 14, 263–274 (2012).
- 4) Sales P. A. Junior., Molina I., Fonseca Murta S. M., Sánchez-Montalvá A., Salvador F., Corrêa-Oliveira R., Carneiro C. M., Am. J. Trop. Med. Hyg., 97, 1289–1303 (2017).
- 5) Morillo C. A., Marin-Neto J. A., Avezum A., et al., N. Engl. J. Med., 373, 1295–1306 (2015).
- 6) Morillo C. A., Waskin H., Sosa-Estani S., et al., J. Am. Coll. Cardiol., 69, 939–947 (2017).
- 7) Molina I., Gomez i Prat J., Salvador F., Trevino B., Sulleiro E., Serre N., Pou D., Roure S., Cabezos J., Valerio L., Blanco-Grau A., Sanchez-Montalva A., Vidal X., Pahissa A., N. Engl. J. Med., 370, 1899–1908 (2014).
- 8) Torrico F., Gascon J., Ortiz L., et al., Lancet Infect. Dis., 18, 419–430 (2018).
- 9) Villalta F., Rachakonda G., Expert Opin. Drug Discov., 14, 1161–1174 (2019).
- 10) Uchiyama N., Kiuchi F., Ito M., Honda G., Takeda Y., Khodzhimatov O. K., Ashurmetov O. A., J. Nat. Prod., 66, 128–131 (2003).
- 11) Uchiyama N., Ito M., Kiuchi F., Honda G., Takeda Y., Khodzhimatov O. K., Ashurmetov O. A., Tetrahedron Lett., 45, 531–533 (2004).
- 12) Suto Y., Kaneko K., Yamagiwa N., Iwasaki G., Tetrahedron Lett., 51, 6329–6330 (2010).
- 13) Suto Y., Nakajima-Shimada J., Yamagiwa N., Onizuka Y., Iwasaki G., Bioorg. Med. Chem. Lett., 25, 2967–2971 (2015).
- 14) Majetich G., Yu J., Org. Lett., 10, 89–91 (2008).
- 15) The synthesis of compounds 4–10 is described in the supplementary material.