Abstract
We have developed efficient synthetic reactions using enamines and enamides carrying oxygen atom substituent on nitrogen, such as N-alkoxyenamines, N,α-dialkoxyenamines, N-alkoxyanamides, and N-(benzoyloxy)enamides. The umpolung reaction by polarity inversion at the β-position of N-alkoxyenamines afforded α-alkyl-, α-aryl-, α-alkenyl-, and α-heteroarylketones by using aluminum reagent as nucleophiles. Furthermore, one-pot umpolung α-phenylation of ketones has been also developed. We applied this method to umpolung reaction of N,α-dialkoxyenamine, generated from N-alkoxyamide to afford α-arylamides. The vicinal functionalization of N-alkoxyenamines has been achieved with the formation of two new carbon–carbon bonds by using an organo-aluminum reagent and subsequent allyl magnesium bromide or tributyltin cyanide. A sequential retro-ene arylation has been developed for the conversion of N-alkoxyenamides to the corresponding tert-alkylamines. The [3,3]-sigmatropic rearrangement of N-(benzoyloxy)enamides followed by arylation afforded cyclic β-aryl-β-amino alcohols bearing a tetrasubstituted carbon center. The resulting products were converted into the corresponding sterically congested cyclic β-amino alcohols, as well as the dissociative anesthetic agent Tiletamine.
1. Introduction
The enamines have been employed as useful substrates, especially for carbon-carbon bond formation, which are among the most important transformations in organic synthesis.1–10) A common route for enamine production is via an acid-catalyzed nucleophilic reaction of ketone or aldehyde species containing an α-hydrogen with secondary amines. Unlike standard alkenes, the carbon-carbon double bond in enamines is polarized due to the presence of a nitrogen atom. Therefore, the carbon at the β-position is nucleophilic, whereas the electron-depleted carbon at the α-position is a latent electrophilic site11–15) (Fig. 1). The enamines can attack electrophiles to form an unstable substituted iminium salt intermediate which then hydrolyzes to afford α-substituted ketone. However, enamines are highly sensitive toward hydrolysis, thereby creating serious difficulties in their experimental handling. In contrast, enamines with an acyl group on the nitrogen atom, i.e., enamides are shelf-stable enamine surrogates that display a diminished enaminic reactivity but an increased stability as the result of the N-electron-withdrawing substituent present on the nitrogen atom.
α,β-Disubstituted amine derivatives are obtained from enamides by bifunctionalization to the α- and β-positions.16,17) Enamides can react with electrophiles to form reactive N-acyliminium intermediates, which then readily react with nucleophiles to give α,β-disubstituted amine derivatives. This approach has been used to synthesize a wide variety of nitrogen-cantaining molecules, which are common motifs in natural products and biologically active compounds.
The substituent R on the nitrogen of the enamines and enamides shown in Fig. 1 is most often an alkyl group. We were interested in the effect of nitrogen substituents on the reactivity of enamines and enamides, and started to develop reactivity of N-alkoxyenamines and enamides with oxygenated functional groups on nitrogen. This review presents our studies on development of efficient synthetic reactions using enamines and enamides carrying an oxygen atom substituent on nitrogen, such as N-alkoxyenamines, N,α-dialkoxyenamines, N-alkoxyenamides, and N-(benzoyloxy)enamides (Fig. 2). The presence of a relatively lower energy N–O bond gives these compounds ability to afford various types of products via cleavage of the N–O bond and formation of new stronger bonds.
2. Nucleophilic α-Alkylation and α-Arylation of Carbonyl Compounds by Polarity Inversion of N-Alkoxyenamines
At first, we introduce the reactivity of N-alkoxyenamines 2 (X = OR). Generally, enamine 2 (X = alkyl), prepared from carbonyl compound 1 and sec-amine, reacts with an electrophile to give a 2-substituted ketone 3 (Chart 1). We anticipated that enamine 2 carrying an oxygen atom substituent (X = OR) would generate the corresponding α-carbonyl carbocation equivalent B by coordination with a Lewis acid followed by N–X bond cleavage of the resulting complex A. The subsequent desired reaction with a nucleophile would occur at an electrophilic α-carbon atom to form the imine C, which is sufficiently unstable to undergo hydrolysis on aqueous work-up to give 4. This reaction corresponds to the polarity inversion reaction at the α-position of carbonyl compounds. The umpolung reaction of the α-carbon on a carbonyl structure is an attractive reaction because it allows the direct introduction of various types of substituents into the α-position through the use of nucleophiles.

Chart 1. Umpolung Strategy Using an Enamine Intermediate
This method is remarkable in its ability to deliver α-aryl ketones, which are normally difficult to prepare using the reaction of an enamine with electrophiles. Organoaluminum reagents were chosen as the nucleophile because we expected that the desired reaction with a nucleophile such as trialkylaluminum, which has Lewis acidic character, could proceed efficiently by sequential coordination, N–O bond cleavage, and addition of the nucleophile (complex D in Chart 1).
The umpolung reaction of the α-carbon atom on the carbonyl structure is an attractive reaction because it allows the direct introduction of various types of substituents into the α-position through the use of nucleophiles. To the best of our knowledge, less is known about the umpolung reaction at the α-position of the carbonyl group except for the use of α-halogenated carbonyl compounds.18–26) There are a few reports of the umpolung reaction of enamine derivatives such as vinyl azides,27) N-sulfonylazoalkenes,28) and enammonium salts with an indolo[2,3-a]quinolizine structure.29) Most umpolung reaction of these enamine derivatives have been carried out using nucleophilic alkylation reagents; however, the umpolung arylation reactions of enamines have been much less investigated.28)
The α-alkylation,30) arylation,30) alkenylation,31) and heteroarylation32) of carbonyl compounds using a nucleophile were investigated as follows.
2-1. Nucleophilic α-Alkylation of Ketones30)Our investigations of this new umpolung strategy focused on the reaction of N-alkoxyenamine 2a bearing a cyclohexene ring with Et3Al. N-alkoxyenamine 2a was prepared from cyclohexanone (1a) and isoxazolidine. As a result, we found optimized reaction conditions as follows.
The solution of cyclohexanone in CH2Cl2 was treated with isoxazolidine in the presence of MgSO4 for 16 h. Then, the reaction mixture was filtered and the filtrate was used for the following reaction without evaporate-on of the solvent. Et3Al was added directly to the filtrate to give 4a in 76% yield (Chart 2).

Chart 2. Enamine Formation and the Umpolung Reaction
Instead of Et3Al, use of Et2Zn, RLi, and RMgX were examined. However, attempts with Et2Zn led to complete recovery of the cyclohexanone, while the reaction with highly reactive organometallic reagents such as alkyllithium and Grignard reagent gave a complex mixture.
With the optimal reaction conditions for the umpolung reaction identified, we next investigated the umpolung alkylation of various ketones (Table 1). At first, we evaluated the reaction of cyclohexanone derivatives 1a–c with several commercially available trialkylaluminums. The expected reaction of cyclohexanone enamine 2a, formed in situ, with iBu3Al proceeded to afford 4b in 69% yield (entry 2). The 4-phenylcyclohexanone enamine 2b prepared from 1b, successfully underwent the umpolung alkylation to provide 4c–e in more than 70% yield (entries 3–5). In the case of 2-methylcyclohexanone (1c), 2-ethyl-6-methylcyclohexanone (4f) was obtained as a result of regioselective formation of trisubstituted enamine 2c as an intermediate.
Table 1. Umpolung Alkylation of Various Ketones
We turned our attention to investigating the umpolung reaction using acyclic ketones. The 5-nonanone (1d) provided good yield of the corresponding ethylated product 4g (entry 7). Similarly, acyclic ketones 1e and 1f gave 4h and 4i in moderate yields (entries 8 and 9). In the case of ketone 1g, 4j was regioselectively obtained via a more stable enamine (entry 10).
To clarify the reaction pathway, we investigated the one-pot double nucleophilic alkylation of N-alkoxyenamine 2a (Chart 3). The treatment of enamine 2a with Et3Al followed by reaction with allylmagnesium bromide gave dialkylated aminoalcohol 6 in 46% yield. This result indicated that this umpolung reaction would proceed via intermediary imine 5, generated by formation of complex E and subsequent umpolung α-alkylation through ring-opening of isoxazolidine. Finally, addition of allylmagnesium bromide to imine 5 gave the amine 6. Instead of allylation, treatment of 5 with water during workup afforded 2-alkylated ketone 4a.

Chart 3. One-Pot Double Nucleophilic Reaction
Encouraged by these results, this umpolung reaction was extended to α-arylation (Table 2). We expected that this type of umpolung reaction would be able easily to furnish α-arylketones.
Table 2. Umpolung Arylation of Various Ketones
We examined the arylation of cyclohexanone enamine 2a with various types of triarylaluminums.33) The expected reaction using Ph3Al proceeded smoothly to give α-phenylated product 7a in 60% yield (entry 1). The scope of triarylaluminums used is exemplified (entries 2–5).
The electronic nature of the substituents on the benzene ring had little effect on the reaction efficiency. The higher yields of desired α-arylketones were observed when 5-nonanone enamine 2d was subjected to α-arylation reaction (entries 6–10). We next examined α-arylation of various types of ketone enamines with (4-MeOC6H4)3Al. The cyclohexanone derivatives 1b and 1c gave stereoselectively the corresponding α-arylated products 7k and 7l in 68 and 60% yields, respectively (entries 11 and 12). The reaction of acyclic ketones 1h and 1i gave desired products 7m and 7n in good yields (entries 13 and 14). However, the ketones 1e and 1g bearing a phenyl group gave the products 7o and 7p in diminished yield (entries 15 and 16). Interestingly, unsymmetrical aliphatic ketone enamines 1j–l underwent regioselective α-arylation predominantly to yield 7q–s (entries 17–19). It is worthy to note that functional groups such as the alkenyl and cyano groups were tolerated during this reaction (entries 18 and 19).34–40)
2-3. Nucleophilic α-Alkenylation of Ketones31)We next confirmed that this method is suitable for synthesis of β,γ-unsaturated ketones. β,γ-Unsaturated ketones are an important molecular scaffold because of their presence in biologically active and natural compounds, including the histone deacetylase (HDAC) inhibitor trichostatin A,41–45) antitumor agent lactimidomycin,46–48) antifungal agent khafrefungin,49–51) and anticancer drug taxol.52–54) In addition, β,γ-unsaturated ketones also serve as versatile intermediates in organic synthesis because of the well-studied chemistry of carbonyls and olefins.55–60)
A reliable method for the synthesis of β,γ-unsaturated ketones is widely known43,61–69) as a three-step synthesis and consists of a cross-aldol reaction of different aldehydes, Wittig olefination of β-hydroxy aldehyde, and oxidation of homoallylic alcohol. In contrast, less is known about the α-alkenylation of ketones via enamines,70,71) although the chemistry of enamine has been widely studied.
We first investigated the umpolung β-alkenylation of N-alkoxyenamine 2j (Chart 4), where 2j was prepared in situ from 2-hexanone (1j) and isoxazolidine. Alkenyl aluminum reagent 8a was prepared in situ by hydroalumination of 1-pentyne with diisobutylaluminium hydride (DIBAL)-H.72–74) After the formation of 2j from 1j and isoxazolidine in the presence of MgSO4, the reaction of 2j with alkenyl aluminum reagent 8a (3 equivalent (equiv.)) proceeded smoothly at 0 °C to afford α-alkenyl ketone 9ja in 62% yield.

Chart 4. Nucleophilic β-Alkenylation Using N-Alkoxyenamine
We next proceeded to evaluate the scope of this reaction by investigating the introduction of various alkenyl group to N-alkoxyenamine 1j (Table 3). The umpolung reaction of 2j with sterically hindered (tert-butyl group) alkenyl aluminum reagent 8b also proceeded to afford β,γ-unsaturated ketone 9jb in 59% yield (entry 1). Alkenyl aluminum reagents 8c (R = cPropyl) and 8d (R = cHexyl) gave the corresponding α-alkenylated products 9jc and 9jd in moderate yields (entries 2 and 3). The use of alkenyl aluminum reagent 8e bearing a benzyl group (R = CH2Ph) gave β,γ-unsaturated ketone 9je in moderate yield (entry 4). For the alkenyl aluminum reagents 8f and 8g carrying extended alkyl chains, α-alkenylated products 9jf and 9jg were obtained in low yield (entries 5 and 6).
Table 3. Nucleophilic β-Alkenylation of
N-Alkoxyenamine Using Various Alkenyl Aluminum Reagents
 |
|---|
| entry | R | product | yield (%) |
|---|
| 1 | tBu (8b) | 9jb | 59 |
| 2 | cPr (8c) | 9jc | 44 |
| 3 | cHexyl (8d) | 9jd | 51 |
| 4 | CH2Ph (8e) | 9je | 56 |
| 5 | CH2CH2Ph (8f) | 9jf | 28 |
| 6 | CH2CH2CH2CH2Cl (8g) | 9jg | 30 |
The scope of different ketones in the synthetic protocol was then explored using several alkenyl aluminum reagents (Table 4). The reaction of acyclic ketones 1k and 1l, carrying terminal alkene and nitrile, with 8a proceeded smoothly to afford the corresponding β,γ-unsaturated ketones 9ka and 9la in yields of 71 and 55%, respectively (entries 1 and 2). Cyclic ketones such as cyclohexanone (1a) and cyclododecanone (1m) also gave the corresponding α-alkenylated products 9aa and 9ma in moderate to good yields (entries 3 and 4). We also examined the use of cyclic ketones with two different alkenyl aluminum reagents, which also afforded the corresponding expected products 9ad and 9me in moderate to good yields (entries 5 and 6).
Table 4. Nucleophilic β-Alkenylation of Various
N-Alkoxyenamines Using Alkenyl Aluminum Reagents
Through further investigation, we developed a one-pot umpolung arylation reaction involving enamine formation. We expected that triarylaluminum would preferentially react with in situ-derived N-alkoxyenamine 2 over the ketone 1 owing to the steric hindrance caused by the bulky substituents of the triarylaluminum reagent. The excess amount of triarylaluminum reagents would trap H2O which was generated by the condensation reaction of the ketone 1 and isoxazolidine. We therefore examined a simple one-pot procedure involving enamine formation without MgSO4 and the umpolung arylation reaction to give α-arylketone 7 (Chart 5).

Chart 5. One-Pot Strategy of Umpolung α-Arylation of Ketones
To a solution of 5-nonanone (1d) and isoxazolidine was added triphenylaluminum reagent prepared from AlCl3 and phenylmagnesium chloride in tetrahydrofuran (THF). The desired product 7f was obtained in 78% yield (Chart 6). The reaction of other ketones 1k and 1b gave the desired products 7t and 7u in 81 and 58% yields, respectively. When phenylmagnesium bromide was employed in place of phenylmagnesium chloride, α-bromoalkoxyketones 10a, 10b, and 10c were obtained as by-products.

Chart 6. One-Pot Umpolung α-Arylation of Ketones 1d, 1k and 1b
We have proposed a possible reaction pathway for bromobutoxylated compounds 10 (Chart 7). The bromide ion attacked the 2-position of THF (solvent) which was activated by aluminum reagents to give bromobutoxyaluminum F. Then, the N-alkoxyenamine 2 was reacted with F to afford the α-bromobutoxylated ketone 10. In the case of phenylmagnesium chloride, the corresponding by-products were not obtained, presumably due to the difference in nucleophilicity between bromide ion and chloride ion.

Chart 7. Possible Reaction Pathway for Bromobutoxylated Compounds
To explore the scope of the reaction, we examined one-pot umpolung reaction of various ketones and the phenylated products shown in Fig. 3 were obtained in 51–79% yields.
2-5. α-Heteroarylation of Ketones Using One-pot Method32)We next investigated methods for the efficient construction of α-heteroarylated ketones using one-pot umpolung reaction. Heteroaromatic units are important substructures that can be found in a wide range of both naturally occurring and synthetic organic compounds with important pharmacological activities.76–78)
We described the umpolung α-heteroarylation reaction of ketones with a range of heterocycles, including thiophene, furan, benzothiophene, benzofuran, and indole.
We initially examined the reaction of 5-nonanone (1d) with tris(2-thienyl)aluminum (11a), which was generated by the transmetalation reaction of commercially available 2-thienyllithium with aluminum chloride (Chart 8). The resulting aluminum reagent 11a was then applied to our previously described method for the umpolung α-arylation reaction of ketones via N-alkenylisoxazolidine. The desired α-thienylated product 12a was obtained in 70% yield.

Chart 8. Thienylation of 5-Nonanone 1d
Encouraged by this result, we proceeded to evaluate the scope of this reaction by investigating the introduction of various thiophenes and furans to 5-nonanone (1d) (Table 5).
Table 5. Umpolung α-Heteroarylation of 5-Nonanone (
1d) with Various Tris(heteroaryl)aluminums
Several heteroarenes were lithiated under standard conditions and transmetalated with aluminum chloride before being added to a solution of 1d and isoxazolidine in dichloromethane. In the cases of the tris(5-substituted 2-thienyl)aluminums, which were derived from thiophenes containing methyl and methoxy groups, the reactions proceeded at the 2-position of thiophene to afford the desired products 12b and 12c in 80 and 65% yields, respectively. The conditions were also applied to tris(5-chloro-2-thienyl) aluminum to allow for the introduction of a 5-chlorothienyl group to 1d. This reaction proceeded smoothly to give the α-heteroarylated product 12d in 73% yield. The reactions of 1d with tris(2-furyl) aluminum and tris(5-methyl-2-furyl) aluminum also proceeded smoothly to give the corresponding α-furylated products 12e and 12f in 64 and 77% yields, respectively.
To further explore the scope of this transformation, we investigated the reactions of various ketones with tris(5-methyl-2-thienyl)aluminum and tris(5-methyl-2-furyl)aluminum (Table 6). When the reaction was applied to unsymmetrical acyclic ketone 1j bearing a methyl group as R1, the α-heteroarylation proceeded in a regioselective manner on the opposite side to that of the methyl group to afford 12g (entry 1). Ketones bearing an olefin 1k or a nitrile 1l were also successfully heteroarylated with tris(5-methyl-2-thiophenyl) aluminum under the standard conditions to give 12h and 12i in 71 and 86% yields, respectively (entries 2 and 3). The reaction conditions were also applicable to cyclic ketones, including cyclohexanone (1a) and cycloheptanone (1o), which gave the α-heteroarylated ketones 12j and 12k in moderate yields (entries 4 and 5). Tris(5-methyl-2-furyl)aluminum also reacted with various N-alkoxyenamines generated from 1j,k,l under these conditions to afford the corresponding α-furylketones 12l–n in moderate yields (entries 6–8).
Table 6. Heteroarylation of Various Ketones
 |
|---|
| entry | substrate | R1 | R2 | X | product | yield (%) |
|---|
| 1 | 1j | Me | nPr | S | 12g | 83 |
| 2 | 1k | Me | Allyl | S | 12h | 71 |
| 3 | 1l | Me | (CH2)2CN | S | 12i | 86 |
| 4 | 1a | –(CH2)4– | S | 12j | 63 |
| 5 | 1o | –(CH2)5– | S | 12k | 60 |
| 6 | 1j | Me | nPr | O | 12l | 62 |
| 7 | 1k | Me | Allyl | O | 12m | 58 |
| 8 | 1l | Me | (CH2)2CN | O | 12n | 58 |
We next investigated the introduction of heteroaryl groups fused to a benzene ring, such as benzothiophene, benzofuran, and 1-methylindole (Table 7). The heteroarylaluminum reagents were prepared according to the same procedure described above for the thienylation reactions. Pleasingly, 5-nonanone (1d) reacted successfully with the aluminum reagents prepared from 2-lithium benzothiophene, 2-lithium benzofuran, and 2-lithium 1-methylindole to give the heteroarylated products as a mixture of the two regioisomers 13 and 14 (entries 1–3). Surprisingly, the umpolung reaction had proceeded not only at the 2-position, but also at the 3-position of these heteroarene systems, despite the lithiation reaction taking place exclusively at their 2-position.79)
Table 7. Introduction of Heteroaryl Groups Fused to a Benzene Ring
 |
|---|
| entry | substrate | R1 | R2 | X | products | yield (%) (13 : 14) |
|---|
| 1 | 1d | nBu | nPr | S | 13a, 14a | 68 (1 : 1) |
| 2 | 1d | nBu | nPr | O | 13b, 14b | 70 (3 : 1) |
| 3 | 1d | nBu | nPr | NMe | 13c, 14c | 60 (1 : 3) |
| 4 | 1j | Me | nPr | NMe | 13d, 14d | 62 (1 : 3) |
| 5 | 1k | Me | Allyl | NMe | 13e, 14e | 60 (1 : 3) |
| 6 | 1l | Me | (CH2)2CN | NMe | 13f, 14f | 73 (1 : 3) |
| 7 | 1a | –(CH2)4– | NMe | 13g, 14g | 63 (1 : 3) |
Furthermore, in the case of the tris(1-methyl-2-indolyl) aluminum reagent, the reaction occurred preferentially at the 3-position. Various ketones 1a,j,k,l were also subjected to the reaction with tris(1-methyl-2-indolyl)aluminum (entries 4–7). The use of unsymmetrical linear ketones 1j,k,l also gave the heteroarylated products 13d–f and 14d–f as 1 : 3 mixtures of the regioisomers in moderate yields (entries 4–6). Cyclohexanone (1a) also reacted in a similar manner with tris(1-methyl-2-indolyl) aluminum to afford the corresponding α-heteroarylated cyclohexanones 13g and 14g as a 1 : 3 mixture of regioisomers in 63% yield (entry 7).
Based on these results, we have proposed a plausible mechanism for the formation of 13 and 14 (Chart 9). Several reports have been published concerning the reaction of aryl borates with electrophiles, where the reaction does not proceed in a regioselective manner at the ipso-position.80–86) Mayr and colleagues87) reported that the presence of a trifluoroborate group at the 2-position of the indole effectively enhanced the nucleophilicity of the carbon at the 3-position, and demonstrated that the vicinal-activation of the indole with a trifluoroborate group was much more effective than ipso-activation. In the case of the indolylaluminum reagent, the presence of aluminum at the 2-position of N-methylindole may have enhanced the nucleophilicity of its 3-position, causing the reaction to occur preferentially at the 3-position of the indole. The aluminum ate complex 16 would be generated from the lithium chloride formed in situ by the reaction of the corresponding indolyl lithium with aluminum chloride. The aluminum ate complex 16 would react at the 3-position with N-alkoxyenamine 2 to form the Wheland intermediate G, and the hydride shift of intermediate G would give intermediate H, which would subsequently release aluminum to generate the 3-substituted indole 14 (Chart 9, Eq. (1)). The 2-substituted indole 13, however, would be obtained as a result of reaction at the ipso-position of the 2-indolylaluminum reagent (Chart 9, Eq. (2)).

Chart 9. Proposal Reaction Pathways for the Formation of 3-Substituted Indole 14 and 2-Substituted Indole 13
3. Vicinal Functionalization of N-Alkoxyenamines88)
Vicinal functionalization of enamides by two carbon–carbon bond formations has received much attention.89) In contrast, less is known about the vicinal functionalization of enamines involving two carbon–carbon bond formations due to the instability of both the enamine substrate and the imine intermediate. Therefore, nucleophilic addition to the imine intermediate is efficient when it occurs in an intramolecular manner, and cyclic compounds such as 2,3,4-trisubstituted tetrahydroquinolines,90,91) 1,3-diaminotetralines,92) and other cycloadducts93–95) are obtained by vicinal functionalization of enamines (Chart 10, Eq (1)). The aforementioned approaches are generally achieved by using a combination of an electrophile and a nucleophile.

Chart 10. Difunctionalization of Enamine via Two C–C-Bond Formations
We described the successful vicinal functionalization of an N-alkoxyenamine by double nucleophilic reaction using two different nucleophiles (Chart 10, Eq (2)). The development of novel vicinal functionalization using a double nucleophilic reaction clearly offers new perspectives in the chemistry of enamines.
The concept of a double nucleophilic addition to the alkene part of an enamine is outlined in Chart 11. To introduce two carbon-centered nucleophiles to the enamine derivative, it is necessary that the alkene acts as a dication equivalent for successful transformation. Therefore, we envisioned that N-alkoxyenamine 18, which is generated in situ from aldehyde 17 and isoxazolidine, could act as a formal and vicinal dication equivalent, because the umpolung reaction of N-alkoxyenamine 18 could generate imine J by coordination with the first nucleophile (Nu1–M) followed by N–O bond cleavage and simultaneous nucleophilic attack of Nu1–M (Cβ-position) (Chart 10). This imine J would be masked in situ by the tethered metal alkoxide to form N,O-acetal K, which would contribute to the stabilization of imine J. The subsequent second addition of a carbon-centered nucleophile (Nu2–M) to imine J could then proceed to afford the α,β-disubstituted amine 19.

Chart 11. Umpolung β-Phenylation and Sequential Nucleophilic Addition to N-Alkoxyenamine 18
To accomplish this kind of vicival introduction of carbon nucleophiles, we examined the phenylation of enamine 18a, prepared in situ from aldehyde 17a, and sequential nucleophilic addition to the resulting imine J (Table 8).
Table 8. Umpolung β-Phenylation and Sequential Nucleophilic Addition of
N-Alkoxyenamine
18a |
|---|
| entry | Nu-M | product | yield (%) | dr |
|---|
| 1 | AllylMgBr | 19a | 78 | 3 : 1 |
| 2 | AllylTMS | 19a | NDa) | — |
| 3 | Et2AlCN | 20a | 56 | 1 : 1 |
| 4 | Bu3SnCN | 20a | 53 | 1.5 : 1 |
| 5 | LiAlH4 | 21a | 82 | — |
a) α-Phenylaldehyde was obtained in 79% yield.
Allylation and cyanation were selected as the second nucleophilic reaction for our initial evaluation, because the products can be easily transformed into valuable derivatives. Treatment of 17a with Ph3Al in the presence of isoxazolidine and subsequent reaction with allylmagnesium bromide gave homoallylamine 19a in 78% yield with a 3 : 1 diastereoselectivity (entry 1). In contrast, the addition of allyltrimethylsilane to imine J, which is generated in situ after α-phenylation of 17a, did not proceed, and α-phenylaldehyde was obtained in 79% yield (entry 2). For the preparation of α-aminonitrile 20a, we introduced a cyano group by sequential phenylation and cyanation. As a result, both diethylaluminum cyanide and tributyltin cyanide gave α-aminonitrile 20a in moderate yields with low diastereoselectivities (entries 3 and 4). Furthermore, the use of LiAlH4 as a second nucleophile after β-phenylation of 18a led to phenethylamine 21a in good yield (entry 5). Thus, sequential umpolung phenylation and nucleophilic addition of N-alkoxyenamine 18a formed in situ from 17a provided homoallylamine 19a, α-aminonitrile 20a, and phenethylamine 21a in moderate to good yields by changing the second nucleophile.
We then proceeded to examine the generality of the umpolung phenylation and sequential allylation or cyanation using a series of N-alkoxyenamines derived from aldehydes (Table 9).
Table 9. Sequential β-Phenylation–Nucleophilic Addition of Various
N-Alkoxyenamines
The corresponding N-alkoxyenamine 18b, prepared from undecanal (17b) and isoxazolidine, was a good substrate for the β-phenylation/allylation and cyanation to give the desired products 19b and 20b in good yields, albeit with unsatisfactory steroselectivities (entries 1 and 2). Both double nucleophilic reactions using enamine 18c, formed in situ from 3-phenylpropionaldehyde (17c), also proceeded smoothly to provide products 19c and 20c in good yields (entries 3 and 4). Sequential β-phenylation/allylation of the corresponding enamines 18d and 18e of aldehydes 17d and 17e containing terminal and internal olefins worked well (entries 5 and 7), while sequential β-phenylation/cyanation of them gave the corresponding products 20d and 20e in moderate yields (entries 6 and 8). Similarly, double nucleophilic reactions of enamines 18f and 18g, prepared in situ from β-branched aldehyde 17f and phenylacetoaldehyde (17g), also worked well (entries 9–12).
Furthermore, the conversion of α-aminonitrile 20g into unnatural α-amino acid 22 was readily achieved by acid hydrolysis of the cyano group (Chart 12).

Chart 12. Conversion of 20g to Unnatural Amino Acid 22
We next examined the sequential β-phenylation and nucleophilic addition of N-alkoxyenamine 2a, which was derived from cyclohexanone (1a) and isoxazolidine (Chart 13). The introduction of allyl or cyano groups to imine L, which was generated by umpolung β-phenylation of enamine 2a, was achieved to give 23 and 24 in moderate yields, using allylmagnesium bromide or tributyltin cyanide, respectively. In addition, the reduction of imine L by LiAlH4 proceeded smoothly to give amine 25 in good yield.

Chart 13. Sequential β-Phenylation–Nucleophilic Addition of N-Alkoxyenamine 2a
4. Nucleophilic α-Arylation of N,α-Dialkoxylamines (N,O-Ketene Acetals)96)
We applied this method to umpolung reaction of N,α-dialkoxylenamie, generated from N-alkoxylamide to afford α-arylamides.
Amides are ubiquitous building blocks and synthetically versatile intermediates for various chemicals and pharmaceuticals, and are also common subunits in natural products.97–104) Therefore, the α-functionalization of amides is an attractive strategy to access a diverse array of amides.105–115) The intermolecular α-arylation of amides is one of the most straightforward approaches for functionalization.116–128)
We investigated the nucleophilic arylation of N,O-ketene acetals, which are generated in situ from N-alkoxyamides. Furthermore, We succeeded in the asymmetric synthesis of α-arylcarboxylic acids by the diastereoselective α-arylation of an N-alkoxyamide bearing a chiral auxiliary.
In general, O-silyl N,O-ketene acetal 26 has been used as a nucleophile for Mukaiyama-type reactions, and it reacts with various electrophiles at the Cα-position129–133) (Fig. 4). We envisioned that N,O-ketene acetal 27, bearing an alkoxy group on the nitrogen atom, would act as a formal electrophile.
The umpolung reaction of N,O-ketene acetals M, generated from N-alkoxyamide 28, could thus be triggered by coordination with a Lewis acidic nucleophile (Chart 14). Subsequent N–O bond cleavage and nucleophilic addition at the Cα-position would form imidate O via N. Hydrolysis of imidate O by workup would afford α-substituted amide 29, with the nucleophile at the α-position via an umpolung process.

Chart 14. Reactivity of N,O-Ketene Acetals M Generated from 28
We initially investigated the nucleophilic phenylation of O-silyl N,O-ketene acetal 32,134–137) which was generated in situ by the reaction of N-alkoxyamide 30a with a silylating agent and an amine base through a two-step, one-pot process.138,139) As a result, we found optimized reaction conditions as follows. The silylation and nucleophilic phenylation of N-alkoxyamide 30a proceeded smootnly in the presence of TBSOTf (2 equiv.), iPr2NEt (4 equiv.), and Ph3Al (1 equiv.) to give α-phenyl amide 31aA (13%) and tert-butyldimethylsilyl (TBS)-protected amide 31aB (68%). Treatment of 30a with TBSOTf, iPr2NEt, and Ph3Al followed by desilylation with tetrabutylammonium fluoride (TBAF)-AcOH gave only 31aA in 74% yield (Chart 15).

Chart 15. Nucleophilic Phenylation of the N,O-Ketene Acetal 32 Generated in Situ from 30a
To demonstrate the utility of α-phenylated amide 31aA, several transformations were examined (Chart 16). Removal of the 3-hydroxypropylamine moiety by acidic hydrolysis afforded carboxylic acid 33 in good yield. Moreover, the amide moiety of 31aA could be used for the construction of 5,6-dihydro-4H-1,3-oxazine 34.

Chart 16. Transformations of 31aA
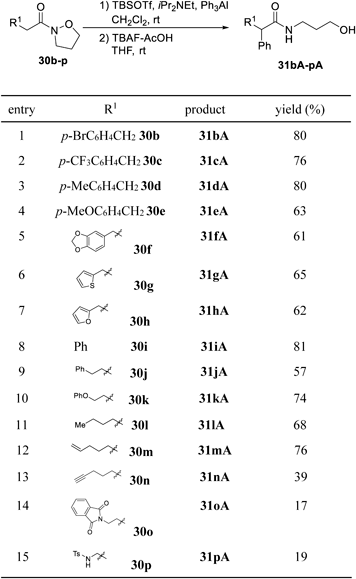
With the optimal conditions for the umpolung reaction in hand, we investigated the nucleophilic phenylation of N,O-ketene acetals generated in situ by treatment of the corresponding N-alkoxyamides 30b–p with TBSOTf and iPr2NEt, followed by deprotection of the TBS group with TBAF (Table 10). The umpolung phenylation of 30b–f with p-bromo, p-trifluoromethyl, p-methyl, p-methoxy, or methylenedioxy groups on the benzene ring proceeded smoothly at ambient temperature to give desired products 31bA–fA in good yields (entries 1–5). The reaction could also be applied to N-alkoxyamides bearing heteroaromatic rings. For example, the umpolung phenylation of 30g and 30h proceeded under the optimized conditions to give corresponding products 31gA and 31hA in moderate yields (entries 6 and 7). In addition, reactions of one-carbon dehomologated N-alkoxyamide 30i and homologated substrate 30j, as well as phenyl ether, alkyl- or olefin-bearing substrates 30k, 30l, and 30m, were also successful (entries 8–12). In contrast, the use of alkyne-, phthalimide-, or tosylamine-bearing substrates 30n, 30o, and 30p in our sequential reaction afforded α-phenylated products 31nA, 31oA, and 31pA in lower yields (entries 13–15).
Table 10. Substrate Scope for Nucleophilic Phenylation of
N,
O-Ketene Acetals Generated
in Situ from
30b–
pTo elucidate the reaction pathway, we confirmed the formation of N,O-ketene acetal 32B under the optimized conditions (Chart 17). When the reaction of N-alkoxyamide 30a with TBSOTf and iPr2NEt was carried out in CDCl3, the olefinic proton signal of 32B in the 1H-NMR spectrum at δ = 4.49 (t, J = 7.0 Hz, 1H) was indicative of a single regioisomer (eq. (1)).140) Subsequent nucleophilic phenylation of 32B with triphenylaluminum proceeded smoothly, and gave α-phenylated product 31aA in 54% yield following the deprotection of the TBS group, indicating that the umpolung reaction proceeded via the in situ formation of N,O-ketene acetal 32B (eq. (1)). To demonstrate the effect of isoxazolidine, we examined the sequential reaction using various amides under the optimized conditions. The use of N-alkoxyamide 35, which bears a tetrahydro-2H-1,2-oxazine instead of isoxazolidine, gave α-phenylated product 36 and TBS derivative 37 in 54% combined yield (eq. (2)), indicating that the isoxazolidine moiety was more suitable for nucleophilic phenylation than tetrahydro-2H-1,2-oxazine. Notably, the sequential reaction of pyrrolidine amide 38 did not occur (eq. (3)), indicating that the adjacent nitrogen and oxygen atoms were critical for this umpolung reaction.

Chart 17. Control Experiments
We next focused on the diastereoselective nucleophilic phenylation of N,O-ketene acetal 40B derived from N-alkoxyamide 39 bearing (+)-benzopyranoisoxazolidine141–145) as the chiral auxiliary (Chart 18). The sequential reaction of 39 with TBSOTf, iPr2NEt, and triphenylaluminum gave α-phenylated product 41a in 10% yield with dr = 1 : 1, and TBS-protected product 42aB in 40% yield with dr = 2 : 1. Interestingly, the use of TESOTf in the diastereoselective α-phenylation led to the α-phenylated product 41a in 39% yield with slightly higher diastereoselctivity (dr = 6 : 1). To confirm the formation of N,O-ketene acetal 40C, the reaction of N-alkoxyamide 39 with TESOTf and iPr2NEt was carried out in CDCl3. However, formation of 40C was not observed by 1H-NMR, indicating that 40C was relatively unstable in the presence of iPr2NEt·TfOH. Therefore, to prepare N,O-ketene acetal 40C in the absence of iPr2NEt·TfOH, we further investigated the diastereoselective nucleophilic phenylation of N,O-ketene acetal 40C which was prepared by an alternative method.

Chart 18. Diastereoselective Nucleophilic Arylation of N,O-Ketene Acetals 40B, 40C under TBSOTf- or TESOTf-iPr2NEt-Ph3Al Systems
Pleasingly, we found that triethylsilylation of the lithium enolate resulted in the formation of (Z)-N,O-ketene acetal 40C146) (Chart 19). Subsequent nucleophilic phenylation of 40C with triphenylaluminum proceeded effectively to give α-phenylated product 41a and triethylsilylated product 42aC, each as single diastereomers (>95% de). The conversion of 42aC to 41a was easily achieved without racemization by addition of 4M HCl.147–150) The chiral amino alcohol moiety of 41a was easily removed by hydrolysis under acidic conditions to afford enantioenriched (R)-2,3-diphenylpropionic acid (33).151)

Chart 19. Diastereoselective Nucleophilic Phenylation of N,O-Ketene Acetal 40C
Next, we examined the scope of the reaction with a variety of triarylaluminum reagents (Table 11). The introduction of various aryl groups, including p-methoxy, p-methyl, and m,p-dimethoxy phenyls, and 2-methyl furan, onto N,O-ketene acetal 40C was achieved in a similar manner, albeit in moderate yields, with the exception of the introduction of a p-chlorophenyl group (entries 1–5). The diastereomeric purities of 41b–f were not less than 95% de, as determined by 1H-NMR analysis.
Table 11. Diastereoselective Nucleophilic Arylation of
N,
O-Ketene Acetal
40C5. Sequential Rearrangement/Nucleophilic Arylation of N-Alkoxyenamides and N-(Benzoyloxy)enamides
5-1. Retro-Ene/Arylation of N-Alkoxyenamides152,153)We next focused on development of reactivity of N-alkoxyenamides 44 in the presence of triarylaluminum reagent. We anticipated that Umpolung arylation of N-alkoxyenamides 44 would proceed to yield α-arylated ketones 4, similar to the case of N-alkoxyenamines. However, it was found that treatment of N-alkoxyenamides 44 with triarylaluminum reagent gave tert-alkylamide derivatives 46 as a result of retro-ene reaction followed by addition of an aryl group to the N-acylketimines 45. (Chart 20). The presence of the strongly electron-withdrawing trifluoroacetyl group effectively enhanced the electrophilicity of the imine in this system, and the group could be readily removed following addition reaction to give the corresponding tert-alkylamines 47, which are widely encountered in natural products and pharmaceuticals.154–156)

Chart 20. Retro-Ene Arylation Reaction of N-Alkoxyenamides 44
The reaction of N-methoxyenamide 48a with tris(4-methoxyphenyl)aluminum, was initially selected as a model reaction to optimize the reaction conditions (Table 12). When the reaction was conducted at ambient temperature, it did not provide any of the desired product, and only the starting material was recovered (entry 1).
Table 12. Optimization of the Retro-Ene Arylation Reaction
 |
|---|
| entry | substrate | R | temp (°C) | yield (%) |
|---|
| 49a | 50a–d |
|---|
| 1 | 48a | H | rt | — | — |
| 2 | 48a | H | 50 | 58 | 34 |
| 3 | 48a | H | 90 | 50 | — |
| 4 | 48b | Ph | 50 | 72 | 53 |
| 5 | 48c | 4-CF3C6H4 | 50 | 68 | 67 |
| 6 | 48d | 4-MeOC6H4 | 50 | 30 | 25 |
Pleasingly, however, when the reaction was conducted at 50 °C, the tert-alkylamide 49a and 4-methoxybenzylalcohol (50a) were obtained in 58 and 34% yields, respectively (entry 2). A further increase in the reaction temperature to 90 °C led to a minor reduction in the yield (entry 3). When we used 4-methoxyphenylmagnesium bromide instead of tris-(4-methoxyphenyl)aluminum, a complex mixture was obtained. The N-alkoxyenamides 48b–d were used to investigate the substituent effects of the different alkoxy groups. When N-benzyloxyenamide 48b was used as substrate, the reaction proceeded smoothly to afford 49a and 50b in 72 and 53% yields, respectively (entry 4). The application of the same reaction conditions to substrate 48c bearing an electron-withdrawing 4-trifluoromethylbenzyloxy group provided similar results (entry 5). In contrast, application of the reaction conditions to substrate 48d bearing an electron-donating 4-methoxybenzyloxy group resulted in lower levels of reactivity and a poorer yield (entry 6).
With an optimized procedure in hand, we proceeded to examine the scope of the reaction by reacting 48b with a range of different triarylaluminum reagents (Table 13).
Table 13. Sequential Retro-Ene Arylation Reactions of a Variety of Different
N-Alkoxyenamides
Use of the triphenylaluminum prepared in situ gave similar results to those observed above for the tris(4-methoxyphenyl)aluminum reagent (entry 1). In contrast, use of the commercially available triphenylaluminum reagent led to a significant reduction in the yield of amide 49b (entry 2). The commercially available triphenylaluminum reagent with MgBr2 gave the product in 50% yield. These results demonstrated that the magnesium salt played an important role in this reaction. The tris(p-tolyl)aluminum and tris(4-fluorophenyl)aluminum reagents also performed well under the optimized conditions to give the desired amides 49c and 49d in relatively good yields (entries 3 and 4, respectively). Various cyclic and acyclic N-alkoxyenamides were then investigated under the optimized reaction conditions further to expand the scope of the reaction (entries 5–11). To evaluate the impact of ring size of cyclic enamides, a substrate bearing a cyclopentenyl group 48e and a substrate bearing a cycloheptenyl group 48f were subjected to the optimized reaction conditions (entries 5 and 6, respectively). When the substrate bearing a cyclopentenyl group was subjected to the reaction conditions, the desired product was obtained in 45% yield following an extended reaction time of 15 h (entry 5). In contrast, the substrate bearing a cycloheptenyl group gave the corresponding amide in 70% yield following a reaction time of only 3 h (entry 6). The reactions of the enamides 48g–i, which contained substituents on their cyclohexene rings, were also examined (entries 7–9). The results revealed that certain functional groups were well-tolerated under the optimized reaction conditions, such as an ethyl ester and an acetal. Compared with 48b, the linear benzyloxyenamides 48j and 48k required extended reaction times to afford the desired amides in moderate yields (entries 10 and 11).
Various reaction conditions were also examined in an attempt to develop a deeper understanding of the reaction and elucidate the mechanism of the reaction. When the alkoxyenamide 48b was treated with magnesium bromide at 50 °C, the N–O bond was cleaved to give enamide 51 and benzaldehyde (52) in 68% and 35% yields, respectively (Chart 21, eq. (1)).

Chart 21. The Reaction under Various Reaction Conditions
When the reaction was conducted in the absence of magnesium bromide, enamide 51 was obtained in a much lower yield of only 7%, with 68% of the starting material also being recovered. These results suggest that the magnesium salt was critical to the retro-ene step of the reaction, and that enamide 51 could be an intermediate in this reaction. Interestingly, however, when enamide 51 was subjected to the optimized conditions, no reaction was observed (Chart 21, eq. (2)). Deuterium labeling experiments were also conducted to investigate the mechanism of this transformation. The deuterated alkoxyenamide 53 was treated with tris(4-methoxyphenyl)aluminum that had been generated in situ at 50 °C and gave the β-deuterated amide 54 in 50% yield (Chart 21, eq. (3)). On the basis of these results, we have proposed a plausible reaction mechanism, as shown in Chart 22. Although the role of the magnesium salt remains unclear, it is envisaged that the magnesium would coordinate with the oxygen atoms of the carbonyl and alkoxy groups to inhibit the free rotation of alkoxy groups and effectively fix the conformation in a manner that favors the retro-ene reaction. The N–O bond would then be cleaved, followed by a 1,5-hydrogen shift to give the N-acylimine 56 and benzaldehyde (52). The retro-ene reaction with concomitant N–O bond cleavage would proceed smoothly because of the formation of a strong C=O bond at the expense of the energy required to break the weak N–O bond.157,158) The aryl groups would then add to the N-acylimine immediately before the occurrence of any tautomerization to the corresponding enamide. The reaction of enamide 48 with triarylaluminum gave amide 49 in low yield even in the absence of the magnesium salt (Table 13, entry 2), but α-arylated ketone such as 4 (Chart 20) was not obtained. It suggests that the arylaluminum also participates in the coordination.

Chart 22. Plausible Reaction Pathway
The benzaldehyde (52) also reacted with triarylaluminum to give the alcohol 50. To complete this work, we proceeded to investigate the removal of the trifluoroacetyl group to afford the corresponding tert-alkylamine 58. When the trifluoroacetamide 49b was treated with NaOH in methanol, the tert-alkylamine 58 was obtained in 91% yield (Chart 23). The cleavage conditions were also successfully applied to trifluoroacetamide 49h, which contained an acetal-protecting group, to give the desired tert-alkylamine product 59 in good yield.

Chart 23. Removal of the Trifluoroacetyl Group
N-Alkoxyenamines and N-alkoxyenamides can be represented by the common structure P (Chart 24). The reaction of N-alkoxyenamines 2 (P: R3 + R4 = CH2CH2) with triarylalminum gave α-aryl carbonyl compounds 4 as a result of umpolung α-arylation, while in the case of N-alkoxyenamides 44 (P: R3 = COCF3), tert-alkylamides 49 were obtained by retro-ene reaction followed by addition of aryl group to imines 56.

Chart 24. Comparison of Reactivity between N-Alkoxyenamine 2 and N-Alkoxyenamide 44
Since N-alkoxyenamines 2 have a cyclic alkoxyamine (isoxazolidine), the reaction of N-alkoxyenamines 2 with triarylaluminium proceeded via the complex Q to give 4.
On the other hand, two possible conformers S and T exist for N-alkoxyenamides 44 because rotation of the N–O bond occurs due to the acyclic structure. The fact that 49 was obtained as a product instead of 4 indicates that the reaction proceeded predominantly via the stable chelating conformer T.
5-3. [3,3]-Sigmatropic Rearrangement/Arylation of N-(Benzoyloxy)enamides159)When N-acyloxyenamide was used instead of N-alkoxyenamide, it was found that [3,3]-sequential sigmatropic rearrangement/arylation proceeded to give β-amino alcohol. β-Amino alcohols are an important group of building blocks that have been used extensively for the preparation of a wide range of complex molecules, including natural products and biologically active compounds.160–162) Although a variety of different methods have been developed for the preparation of β-amino alcohols, the synthetic methodologies for β-aryl-β-amino alcohols bearing a tetrasubstituted carbon center are relatively scarce.
We provided a new process for the synthesis of β-substituted β-amino alcohol derivatives 62 bearing a tetrasubstituted carbon center using sequential [3,3]-sigmatropic rearrangement/nucleophilic addition of N-(benzoyloxy)enamides 60 (Chart 25). Notably, this new process provides facile access to a series of sterically congested β-amino alcohols 63, as well as the dissociative anesthetic agent Tiletamine.

Chart 25. [3,3]-Sigmatropic Rearrangement of N-(Benzoyloxy)enamides
We initially examined the reaction of N-(benzoyloxy)enamide 60a, which was prepared from cyclohexanone oxime via a two-step sequence, with various organoaluminum reagents (Table 14).
Table 14. Optimization of the Sequential Using an Aluminum Reagent
 |
|---|
| entry | aluminum reagent | product: yield (%) | cis/trans |
|---|
| 1 | None | 64: 88 | — |
| 2 | Ph3Al (R = Ph) | 62aA: 74 | 9.5 : 1 |
| 3 | PhAlMe2 (R = Me) | 62aA: 67 | 4.5 : 1 |
| 4 | PhAlEt2 (R = Et) | 62aA: 65 | 7.5 : 1 |
Heating a solution of N-(benzoyloxy)enamide 60a in refluxing tetrahydrofuran for three hours gave α-benzoyloxy ketone 64 in 88% yield (entry 1). Encouraged by this result, we proceeded to investigate the sequential [3,3]-sigmatropic rearrangement/phenylation reaction of 60a in the presence of an organoaluminum reagent. The reaction of 60a with triphenylaluminum in refluxing tetrahydrofuran proceeded smoothly to give the β-phenyl-β-amino alcohol derivatives 62aA as a separable 9.5 : 1 mixture of cis/trans isomers in a combined yield of 74% (entry 2). Dimethyl(phenyl)aluminum and diethyl(phenyl)aluminum also worked well in this reaction to give the desired product 62aA in 67% and 65% yields, respectively, albeit with lower diastereoselectivities than triphenylaluminum (entries 3 and 4).163)
With the optimal conditions in hand for the sequential reaction, we proceeded to examine the scope of the reaction with a range of different triarylaluminum reagents (Table 15).
Table 15. Sequential Reaction of Several
N-Benzoyloxyenamides with Ar
3Al
 |
|---|
| entry | substrate | aluminum reagent | product | yield (%) | cis/trans |
|---|
| 1 | 60a | (4-MeOC6H4)3Al | 62aB: Ar = 4-MeOC6H4 | 69 | 17.5 : 1 |
| 2 | 60a | (4-MeC6H4)3Al | 62aC: Ar = 4-MeC6H4 | 54 | 10.5 : 1 |
| 3 | 60a | (4-FC6H4)3Al | 62aD: Ar = 4-FC6H4 | 13 | >20 : 1 |
| 4 | 60a | (2-MeOC6H4)3Al | 62aE: Ar = 2-MeOC6H4 | 70 | 10.5 : 1 |
| 5 | 60a | (3,4-(MeO)2C6H3)3Al | 62aF: Ar = 3,4-(MeO)2C6H3 | 51 | >20 : 1 |
| 6 | 60b | Ph3Al | 62bA: Ar = Ph | 50 | 2 : 1 |
| 7 | 60c | Ph3Al | 62cA: Ar = Ph | 41 | >20 : 1 |
The results show that triarylaluminum reagents containing an electron-donating group such as tris(4-methoxyphenyl) aluminum and tris(4-methylphenyl)aluminum perform well under the optimized reaction conditions to give the desired products 62aB and 62aC in moderate to good yields with high diastereoselectivities (entries 1 and 2). However, a triarylaluminum reagent containing a halogen atom (e.g., (4-FC6H4)3Al) performed poorly giving the desired product 62aD in only 13% yield (entry 3). Notably, triarylaluminum reagents bearing a methoxy group at the ortho position of their phenyl ring (e.g., (2-MeOC6H4)3Al) or two methoxy groups on their phenyl ring (e.g., 3,4-(MeO)2C6H3)3Al) also performed well to give the corresponding products 62aE and 62aF in moderate to good yields with high diastereoselectivities (entries 4 and 5). We subsequently explored the substrate scope of this reaction using triphenylaluminum. To evaluate the effect of the ring size of cyclic N-(benzoyloxy) enamides, enamide 60b with a seven-membered ring and 60c with a five-membered ring were subjected to the optimized reaction conditions. Interestingly, the sequential reaction of 60b produced the desired product 62bA with low diastereoselectivity, while the reaction of 60c afforded cis 62cA as a single diastereomer (entries 6 and 7).
To demonstrate the utility of 62aA, we investigated its conversion into a variety of other interesting compounds (Chart 26).

Chart 26. Various Transformation of 62aA
For example, treatment of 62aA with sodium hydroxide in methanol allowed the simultaneous removal of its trifluoroacetyl and benzoyl groups to afford the β-amino alcohol 65aA in 81% yield. Notably, oxidation of 65aA with potassium chromate in sulfuric acid provided 2-amino-2-phenylcyclohexanone (66), which is the basic pharmacophore of the dissociative anesthetic drug ketamine.164–166) Moreover, cyclization of cis-amino alcohol 65aA with triphosgene furnished the sterically congested oxazolidinone 67 in high yield.
Nucleophilic heteroarylation is an important reaction in synthetic chemistry because heteroaromatic units can be found in a wide range of important biologically active compounds and functional materials.167) We examined the synthesis of β-heteroaryl-β-amino alcohol derivatives via sequential [3,3]-sigmatropic rearrangement/ nucleophilic heteroarylation of N-(benzoyloxy)enamide 60a (Table 16).
Table 16. Optimization for Sequential Rearrangement/2-Thienylation Reaction
 |
|---|
| entry | aluminum reagent | yield (%) | cis/trans |
|---|
| 1 | (2-thienyl)3Al (R = 2-thienyl) | 7 | >20 : 1 |
| 2 | (2-thienyl)AlMe2 (R = Me) | 71 | >20 : 1 |
| 3 | (2-thienyl)AlEt2 (R = Et) | 62 | >20 : 1 |
Introduction of a 2-thienyl group was initially investigated as a model heteroarylation reaction because several thiophene-containing medicines, including Duloxetine,168–173) Tiotropium,174) Clopidogrel,175–177) Prasugrel,178) and Rivaroxaban,178,179) are already widely used in a wide range of clinical applications. Our initial studies of the nucleophilic heteroarylation of the N-(trifluoroacetyl)ketimine intermediate unexpectedly showed that the reaction of N-(benzoyloxy)enamide 60a with tri-2-thienylaluminum under the optimized conditions developed for the sequential rearrangement/nucleophilic phenylation of 60a in Table 16 yielded the desired product 62aG in low yield (entry 1). Pleasingly, however, when the sequential reaction was conducted with dimethyl(2-thienyl)aluminum instead of tri-2-thienylaluminum, the yield of the desired product 62aG dramatically increased (entry 2). Similarly, the use of diethyl(2-thienyl)aluminum afforded 62aG in moderate yield (entry 3).
These results, therefore, demonstrate that dimethyl(2-thienyl)aluminum is superior to tri-2-thienylaluminum for the sequential rearrangement/2-thienylation of 60a with regards to both the yield of the product and atom economy of the reaction. Encouraged by this result, we proceeded to evaluate the scope of this reaction by investigating the introduction of a variety of different heteroaryl groups into the N-(trifluoroacetyl)ketimine intermediate (Table 17).
Table 17. Introduction of a Variety of Different Heteroaryl Groups
For the dimethyl(5-substituted 2-thienyl)aluminum reagents, which were derived from the corresponding 5-substituted thiophenes containing a methyl, methoxy, or chloro group, the reactions proceeded at the 2-position of the thiophene to afford the desired products 62aH and 62aI in good yields, while 62aJ was obtained in a much lower yield. Similarly, the reactions of 60a with dimethyl(2-furyl)aluminum and dimethyl(5- methyl-2-furyl)aluminum proceeded smoothly to give the corresponding α-furylated products 62aK and 62aL in good yields. Further to explore the scope of this transformation, we also examined the introduction of heteroaryl groups fused to a benzene ring, such as benzothiophene and benzofuran. However, desired products 62aM and 62aN were obtained in unsatisfactory yields.
To demonstrate the synthetic value of this developed sequential rearrangement/heteroarylation reaction, we developed an efficient process for synthesis of the dissociative anesthetic agent Tiletamine (Chart 27), which is also classified as an N-methyl-D-aspartate (NMDA) receptor antagonist.180,181) The deprotection of 62aG with sodium hydroxide in methanol afforded β-(2-thienyl)-β-amino alcohol 65aG in good yield. Subsequent N-ethylation of 65aG under reductive amination conditions, followed by Jones oxidation of the resulting alcohol, provided Tiletamine in an overall yield of 35% over six steps from commercially available cyclohexanone oxime (68). The 1H-NMR spectrum of the synthetic Tiletamine hydrochloride salt180,181) was found identical to that of the commercially available Tiletamine hydrochloride provided by Toronto Research Chemicals Inc.

Chart 27. Synthesis of Tiletamine
We have successfully developed an alternative synthetic method for the construction of cyclic β-aryl-β-amino alcohols containing a tetrasubstituted carbon center bearing a nitrogen via sequential [3,3]-sigmatropic rearrangement/nucleophilic arylation of N-(benzoyloxy)enamides.
Conclusion
As part of our research to clarify the effect of substituents on the nitrogen of enamines and enamides on reactivity, we investigated the reactivity of enamines and enamides carrying oxygen atom substituent on nitrogen, such as N-alkoxyenamines, N,α-dialkoxyenamines, N-alkoxyanamides, and N-(benzoyloxy)enamides with trisubstituted aluminum reagents. As a result, we developed the following reactions useful for synthetic organic chemistry.182–187)
-
1)
An efficient umpolung reaction by polarity inversion at the β-position of N-alkoxyenamines afforded α-alkyl-, α-aryl-, α-alkenyl-, and α-heteroarylketones using aluminum reagents as nucleophiles. Furthermore, the efficient one-pot umpolung α-phenylation of ketones via N-alkenylisoxazolidine has been also developed.
-
2)
We applied this method to umpolung reaction of N,α-dialkoxylenamie, generated from N-alkoxylamide to afford α-arylamides.
-
3)
The vicinal functionalization of N-alkoxyenamines has been achieved with the formation of two new carbon–carbon bonds, using an organo-aluminum reagent and subsequent allyl magnesium bromide or tributyltin cyanide.
-
4)
A sequential retro-ene arylation reaction has been developed for the conversion of N-alkoxyenamides to the corresponding tert-alkylamines.
-
5) The [3,3]-sigmatropic rearrangement of N-(benzoyloxy)enamides afforded cyclic β-aryl-β-amino alcohols bearing a tetrasubstituted carbon center. The resulting products were converted into the corresponding sterically congested cyclic β-amino alcohols, as well as the dissociative anesthetic agent Tiletamine.
Acknowledgments
The experiments performed herein were carried out at our laboratory (Medicinal Chemistry Labratory) in Kobe Pharmaceutical University, and I would like to express heartfelt gratitude to the Emeritus Prof. T. Naito for his support and encouragement. The following collaborators are appreciated for their invaluable efforts: associate Professor (currently Professor) Dr. M. Ueda, Dr. N. Takeda (2014. 4.~), and Mr. T. Miyoshi (~2014. 3). A number of co-workers detailed in the references are also acknowledged for their hard work, creativity, and inventions. Our work was financially supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and from the Japan Society for the Promotion of Science, to whom the author’s thanks are due.
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2016 Pharmaceutical Society of Japan Award for Divisional Scientific Contribution.
References and Notes
- 1) Mukherjee S., Yang J. W., Hoffmann S., List B., Chem. Rev., 107, 5471–5569 (2007).
- 2) Melchiorre P., Marigo M., Carlone A., Bartoli G., Angew. Chem. Int. Ed., 47, 6138–6171 (2008).
- 3) Bertelsen S., Jørgensen K. A., Chem. Soc. Rev., 38, 2178–2189 (2009).
- 4) Bernadat G., Masson G., Synlett, 25, 2842–2867 (2014).
- 5) Gigant N., Chausset-Boissarie L., Gillaizeau I., Chem. Eur. J., 20, 7548–7564 (2014).
- 6) Dake G. R., Synlett, 23, 814–824 (2012).
- 7) Gopalaiah K., Kagan H. B., Chem. Rev., 111, 4599–4657 (2011).
- 8) Carbey D. R., Org. Biomol. Chem., 6, 3455–3460 (2008).
- 9) Matsubara R., Kobayashi S., Acc. Chem. Res., 41, 292–301 (2008).
- 10) Nugent T. C., El-Shazly M., Adv. Synth. Catal., 352, 753–819 (2010).
- 11) Matsubara R., Kawai N., Kobayashi S., Angew. Chem. Int. Ed., 45, 3814–3816 (2006).
- 12) Terada M., Machioka K., Sorimachi K., Angew. Chem. Int. Ed., 48, 2553–2556 (2009).
- 13) Bekkaye M., Su Y., Masson G., Eur. J. Org. Chem., 2013, 3978–3982 (2013).
- 14) Hashimoto T., Nakatsu H., Takiguchi Y., Maruoka K., J. Am. Chem. Soc., 135, 16010–16013 (2013).
- 15) He L., Zhao L., Wang D.-X., Wang M.-X., Org. Lett., 16, 5972–5975 (2014).
- 16) Bernadat G., Masson G., Synlett, 25, 2842–2867 (2014).
- 17) Bouchet D., Varlet T., Masson G., Acc. Chem. Res., 55, 3265–3283 (2022).
- 18) Hussey A. S., Herr R. R., J. Org. Chem., 24, 843–845 (1959).
- 19) Jones P. R., Young J. R., J. Org. Chem., 33, 1675–1676 (1968).
- 20) Cai J., Nemoto H., Singaram B., Yamamoto Y., Tetrahedron Lett., 37, 3383–3386 (1996).
- 21) Amano T., Yoshikawa K., Sano T., Ohuchi Y., Shiono M., Ishiguro M., Fujita Y., Synth. Commun., 16, 499–507 (1986).
- 22) Malosh C. F., Ready J. M., J. Am. Chem. Soc., 126, 10240–10241 (2004).
- 23) Ohmiya H., Yorimitsu H., Oshima K., J. Am. Chem. Soc., 128, 1886–1889 (2006).
- 24) Früstner A., Martin R., Krause H., Seidel G., Goddard R., Lehmann C. W., J. Am. Chem. Soc., 130, 8773–8787 (2008).
- 25) Lundin P. M., Esquivias J., Fu G. C., Angew. Chem. Int. Ed., 48, 154–156 (2008).
- 26) Lou S., Fu G. C., J. Am. Chem. Soc., 132, 1264–1266 (2010).
- 27) Hassner A., Belinka B. A. Jr., J. Am. Chem. Soc., 102, 6185–6186 (1980).
- 28) Hatcher J. M., Coltart D. M., J. Am. Chem. Soc., 132, 4546–4547 (2010).
- 29) Lukács A., Szabó L., Baitz-Gács E., Bombicz P., Dobó A., Kalaus G., Szántay C., Heterocycles, 48, 2507–2520 (1998).
- 30) Miyoshi T., Miyakawa T., Ueda M., Miyata O., Angew. Chem. Int. Ed., 50, 928–931 (2011).
- 31) Nandi R. K., Takeda N., Ueda M., Miyata O., Tetrahedron Lett., 57, 2269–2272 (2016).
- 32) Miyoshi T., Takeda N., Fukami M., Sato S., Ueda M., Miyata O., Chem. Pharm. Bull., 62, 927–932 (2014).
- 33) Haraguchi K., Kubota Y., Tanaka H., J. Org. Chem., 69, 1831–1836 (2004).
- 34) Zawodny W., Teskey C. J., Mishevska M., Völkl M., Maryasin B., González L., Maulide N., Angew. Chem. Int. Ed., 59, 20935–20939 (2020).
- 35) Huang X., Zhang Y., Zhang C., Zhang L., Xu Y., Kong L., Wang Z.-X., Peng B., Angew. Chem. Int. Ed., 58, 5956–5961 (2019).
- 36) Hyatt I. F. D., Dave L., David N., Kaur K., Medard M., Mowdawalla C., Org. Biomol. Chem., 17, 7822–7848 (2019).
- 37) Nguyen T. N., Setthakarn K., May J. A., Org. Lett., 21, 7837–7840 (2019).
- 38) Silva F. C. S., Van N. T., Wengryniuk S. E., J. Am. Chem. Soc., 142, 64–69 (2020).
- 39) Martins B. S., Kaiser D., Bauer A., Tiefenbrunner I., Maulide N., Org. Lett., 23, 2094–2098 (2021).
- 40) Nguyen T. N., Setthakarn K., May J. A., Org. Lett., 21, 7837–7840 (2019).
- 41) Tsuji N., Kobayashi M., Nagashima K., Wakisaki Y., Koizumi K., J. Antibiot. (Tokyo), 29, 1–6 (1976).
- 42) Fleming I., Iqbal J., Krebs E.-P., Tetrahedron, 39, 841–846 (1983).
- 43) Mori K., Koseki K., Tetrahedron, 44, 6013–6020 (1988).
- 44) Zhang S., Duan W., Wang W., Adv. Synth. Catal., 348, 1228–1234 (2006).
- 45) Cosner C. C., Iska V. B. R., Chatterjee A., Markiewicz J. T., Corden S. J., Löftstedt J., Ankner T., Richer J., Hulett T., Schauer D. J., Wiest O., Helquist P., Eur. J. Org. Chem., 2013, 162–172 (2013).
- 46) Sugawara K., Nishiyama Y., Toda S., Komiyama N., Hatori M., Moriyama T., Sawada Y., Kamei H., Konishi M., Oki T., J. Antibiot. (Tokyo), 45, 1433–1441 (1992).
- 47) Micoine K., Früstner A., J. Am. Chem. Soc., 132, 14064–14066 (2010).
- 48) Li W., Georg G. I., Chem. Commun. (Camb.), 2015, 8634–8636 (2015).
- 49) Mandala S. M., Thornton R. A., Rosenbach M., Milligan J., Garcia-Calvo M., Bull H. G., Kurtz M. B., J. Biol. Chem., 272, 32709–32714 (1997).
- 50) Wakabayashi T., Mori K., Kobayashi S., J. Am. Chem. Soc., 123, 1372–1375 (2001).
- 51) Shirokawa S., Shinoyama M., Ooi I., Hosokawa S., Nakazaki A., Kobayashi S., Org. Lett., 9, 849–852 (2007).
- 52) Wani M. C., Taylor H. L., Wall M. E., Coggon P., McPhail A. T., J. Am. Chem. Soc., 93, 2325–2327 (1971).
- 53) Holton R. A., Somoza C., Kim H. B., Liang F., Biediger R. J., Boatman P. D., Shindo M., Smith C. C., Kim S., Nadizadeh H., Suzuki Y., Tao C., Vu P., Tang S., Zhang P., Murthi K. K., Gentile L. N., Liu J. H., J. Am. Chem. Soc., 116, 1597–1598 (1994).
- 54) Hirai S., Utsugi M., Iwamoto M., Nakada M., Chem. Eur. J., 21, 355–359 (2015).
- 55) Zou Y., Ding C., Zhou L., Li Z., Wang Q., Schoenebeck F., Goeke A., Angew. Chem. Int. Ed., 51, 5647–5651 (2012).
- 56) Martin-Fontecha M., Agarrabeitia A. R., Ortiz M. J., Armesto D., Org. Lett., 12, 4082–4085 (2010).
- 57) Keränen M. D., Kot K., Hollmann C., Eilbracht P., Org. Biomol. Chem., 2, 3379–3384 (2004).
- 58) Hollmann C., Eilbracht P., Tetrahedron Lett., 40, 4313–4316 (1999).
- 59) Mathew J., J. Org. Chem., 56, 713–716 (1991).
- 60) Houk K. N., Chem. Rev., 76, 1–74 (1996).
- 61) Grigalunas M., Ankner T., Norrby P.-O., Wiest O., Helquist P., J. Am. Chem. Soc., 137, 7019–7022 (2015).
- 62) Grigalunas M., Ankner T., Norrby P.-O., Wiest O., Helquist P., Org. Lett., 16, 3970–3973 (2014).
- 63) Huang J., Bunel E., Faul M. M., Org. Lett., 9, 4343–4546 (2007).
- 64) Chieffi A., Kamikawa K., Åhman J., Fox J. M., Buchwald S. L., Org. Lett., 3, 1897–1900 (2001).
- 65) Lou S., Fu G. C., J. Am. Chem. Soc., 132, 5010–5011 (2010).
- 66) Xu T., Hu X., Angew. Chem. Int. Ed., 54, 1307–1311 (2015).
- 67) Trofimov B. A., Schmidt E. Y., Ushakov I. A., Zorina N. V., Skital’tseva E. V., Protsuk N. I., Mikhaleva A. I., Chem. Eur. J., 16, 8516–8521 (2010).
- 68) Trofimov B. A., Schmidt E. Y., Zorina N. V., Ivanova E. V., Ushakov I. A., J. Org. Chem., 77, 6880–6886 (2012).
- 69) Trofimov B. A., Schmidt E. Y., Zorina N. V., Ivanova E. V., Ushakov I. A., Mikhaleva A. I., Adv. Synth. Catal., 354, 1813–1818 (2012).
- 70) Negishi E., Akiyoshi K., Chem. Lett., 1987, 1007–1010 (1987).
- 71) Fujimoto T., Endo K., Tsuji H., Nakamura M., Nakamura E., J. Am. Chem. Soc., 130, 4492–4496 (2008).
- 72) Cottet P., Müller D., Alexakis A., Org. Lett., 15, 828–831 (2013).
- 73) Müller D., Alexakis A., Org. Lett., 14, 1842–1845 (2012).
- 74) Müller D., Tissot M., Alexakis A., Org. Lett., 13, 3040–3043 (2011).
- 75) Miyoshi T., Sato S., Tanaka H., Hasegawa C., Ueda M., Miyata O., Tetrahedron Lett., 53, 4188–4191 (2012).
- 76) Katritzky A. R., “Comprehensive Heterocyclic Chemistry III,” Vol. III, ed. by Ramsden C. A., Scriven E. F., Taylor R. J. K., Elsevier Press, Oxford, 2008, pp. 353–388.
- 77) Katritzky A. R., “Comprehensive Heterocyclic Chemistry III,” Vol. III, ed. by Ramsden C. A., Scriven E. F., Taylor R. J. K., Elsevier Press, Oxford, 2008, pp. 571–623.
- 78) Katritzky A. R., “Comprehensive Heterocyclic Chemistry III,” Vol. III, ed. by Ramsden C. A., Scriven E. F., Taylor R. J. K., Elsevier Press, Oxford, 2008, pp. 931–974.
- 79) The quenching of lithiated 1-methylindole with D2O provided 2-deutero-1-methylindole, see: Lane B. S., Brown M. A., Sames D., J. Am. Chem. Soc., 127, 8050–8057 (2005).
- 80) Molander G. A., Cavalcanti L. N., J. Org. Chem., 76, 7195–7203 (2011).
- 81) Kim J., Movassaghi M., J. Am. Chem. Soc., 133, 14940–14943 (2011).
- 82) Ishikura M., Kato H., Tetrahedron, 58, 9827–9838 (2002).
- 83) Ishikura M., Agata I., Katagiri N., J. Heterocycl. Chem., 36, 873–879 (1999).
- 84) Ishikura M., Terashima M., J. Heterocycl. Chem., 31, 977–980 (1994).
- 85) Levy A. B., Tetrahedron Lett., 20, 4021–4024 (1979).
- 86) Marinelli E. R., Levy A. B., Tetrahedron Lett., 20, 2313–2316 (1979).
- 87) Berionni G., Morozova V., Heininger M., Mayer P., Knochel P., Mayr H., J. Am. Chem. Soc., 135, 6317–6324 (2013).
- 88) Sato S., Takeda N., Miyoshi T., Ueda M., Miyata O., Eur. J. Org. Chem., 2015, 3899–3904 (2015).
- 89) Bernadat G., Masson G., Synlett, 25, 2842–2867 (2014).
- 90) Talukdar S., Chen C.-T., Fang J.-M., J. Org. Chem., 65, 3148–3153 (2000).
- 91) Rong Z., Li Q., Lin W., Jia Y., Tetrahedron Lett., 54, 4432–4434 (2013).
- 92) Dagousset G., Erb W., Zhu J., Masson G., Org. Lett., 16, 2554–2557 (2014).
- 93) Sheldrake H. M., Wallace T. W., Wilson C. P., Org. Lett., 7, 4233–4236 (2005).
- 94) Coskun N., Ma J., Azimi S., Gärtner C., Erden I., Org. Lett., 13, 5952–5955 (2011).
- 95) Anderson E. D., Duerfeldt A. S., Zhu K., Glinkerman C. M., Boger D. L., Org. Lett., 16, 5084–5087 (2014).
- 96) Takeda N., Futaki E., Kobori Y., Ueda M., Miyata O., Angew. Chem. Int. Ed., 56, 16342–16346 (2017).
- 97) Ogliaruso M. A., Wolfe J. F., “Synthesis of Carboxylic Acids, Esters and Their Derivatives,” ed. by Patai S., Rappoport Z., Wiley, New York, 1991, pp. 176–192.
- 98) Peng B., Geerdink D., Maulide N., J. Am. Chem. Soc., 135, 14968–14971 (2013).
- 99) Bechara W. S., Pelletier G., Charette A. B., Nat. Chem., 4, 228–234 (2012).
- 100) Liu C., Achtenhagen M., Szostak M., Org. Lett., 18, 2375–2378 (2016).
- 101) Oda Y., Sato T., Chida N., Org. Lett., 14, 950–953 (2012).
- 102) Lindquist N., Fenical W., Van Duyne G. D., Clardy J., J. Am. Chem. Soc., 113, 2303–2304 (1991).
- 103) Igarashi Y., Futamata K., Fujita T., Sekine A., Senda H., Naoki H., Furumai T., J. Antibiot. (Tokyo), 56, 107–113 (2003).
- 104) Garo E., Starks C. M., Jensen P. R., Fenical W., Lobkovsky E., Clardy J., J. Nat. Prod., 66, 423–426 (2003).
- 105) Kaiser D., Maulide N., J. Org. Chem., 81, 4421–4428 (2016).
- 106) Dander J. E., Garg N. K., ACS Catal., 7, 1413–1423 (2017).
- 107) Ruider S. A., Maulide N., Angew. Chem. Int. Ed., 54, 13856–13858 (2015).
- 108) Hie L., Nathel N. F. F., Shah T. K., Baker E. L., Hong X., Yang Y.-F., Liu P., Houk K. N., Garg N. K., Nature (London), 524, 79–83 (2015).
- 109) Fuentes de Arriba Á., Lenci E., Sonawane M., Formery O., Dixon D., Angew. Chem. Int. Ed., 56, 3655–3659 (2017).
- 110) Morales M. R., Mellem K. T., Myers A. G., Angew. Chem. Int. Ed., 51, 4568–4571 (2012).
- 111) Guo L., Liu Y., Yao W., Leng X., Huang Z., Org. Lett., 15, 1144–1147 (2013).
- 112) Tona V., Torre A., Padmanaban M., Ruider S., González L., Maulide N., J. Am. Chem. Soc., 138, 8348–8351 (2016).
- 113) Lubin H., Tessier A., Chaume G., Pytkowicz J., Brigaud T., Org. Lett., 12, 1496–1499 (2010).
- 114) Lubin H., Dupuis C., Pytkowicz J., Brigaud T., J. Org. Chem., 78, 3487–3492 (2013).
- 115) Kim Y., Chang S., Angew. Chem. Int. Ed., 55, 218–222 (2016).
- 116) Johansson C. C. C., Colacot T. J., Angew. Chem. Int. Ed., 49, 676–707 (2010).
- 117) Bellina F., Rossi R., Chem. Rev., 110, 1082–1146 (2010).
- 118) Zheng B., Jia T., Walsh P. A., Org. Lett., 15, 4190–4193 (2013).
- 119) Lundin P. M., Fu G., J. Am. Chem. Soc., 132, 11027–11029 (2010).
- 120) Shaughnessy K. H., Hamann B. C., Hartwig J. F., J. Org. Chem., 63, 6546–6553 (1998).
- 121) Hama T., Culkin D. A., Hartwig J. F., J. Am. Chem. Soc., 128, 4976–4985 (2006).
- 122) Hama T., Liu X., Culkin D. A., Hartwig J. F., J. Am. Chem. Soc., 125, 11176–11177 (2003).
- 123) Zheng B., Jia T., Walsh P. J., Adv. Synth. Catal., 356, 165–178 (2014).
- 124) Liu C., He C., Shi W., Chen M., Lei A., Org. Lett., 9, 5601–5604 (2007).
- 125) Lu T.-Y., Xue C., Luo F. T., Tetrahedron Lett., 44, 1587–1590 (2003).
- 126) Molander G. A., Traister K. M., Barcellos T., J. Org. Chem., 78, 4123–4131 (2013).
- 127) Peng B., Geerdink D., Farès C., Maulide N., Angew. Chem. Int. Ed., 53, 5462–5466 (2014).
- 128) Shaaban S., Tona V., Peng B., Maulide N., Angew. Chem. Int. Ed., 56, 10938–10941 (2017).
- 129) McGilvra J. D., Unni A. K., Modi K., Rawal V. H., Angew. Chem. Int. Ed., 45, 6130–6133 (2006).
- 130) Gondi V. B., Hagihara K., Rawal V. H., Angew. Chem. Int. Ed., 48, 776–779 (2009).
- 131) Borths C. J., Carrera D. E., MacMillan D. W. C., Tetrahedron, 65, 6746–6753 (2009).
- 132) Gondi V. B., Hagihara K., Rawal V. H., Chem. Commun. (Camb.), 2010, 904–906 (2010).
- 133) Myers A. G., Widdowson K. L., J. Am. Chem. Soc., 112, 9672–9674 (1990).
- 134) Kaiser D., de la Torre A., Shaaban S., Maulide N., Angew. Chem. Int. Ed., 56, 5921–5925 (2017).
- 135) Cupps T. L., Boutin R. H., Rapoport H., J. Org. Chem., 50, 3972–397 (1985).
- 136) Boutin R. H., Rapoport H., J. Org. Chem., 51, 5320–5327 (1986).
- 137) Das R., Kapur M., Chem. Asian J., 10, 1505–1512 (2015).
- 138) For examples of the in situ generation of N,O-ketene acetals from amides by the use of a trialkylsilyl triflate and a base, see: Kobayashi S., Kiyohara H., Yamaguchi M., J. Am. Chem. Soc., 133, 708–711 (2011).
- 139) Downey C. W., Ingersoll J. A., Glist H. M., Dombrowski C. M., Barnett A. T., Eur. J. Org. Chem., 2015, 7287–7291 (2015).
- 140) The C=C group of 32B (Si = TBS) was deduced to have the Z configuration by comparison with the 1H-NMR spectrum of N,O-ketene acetal 43, which was prepared by triethylsilylation of the lithium enolate derived from the one-carbon homologated substrate 30j. This structure was confirmed by 1H-NMR (olefinic H: δ = 4.49 ppm (t, J = 7.0 Hz, 1H)) and NOESY experiments (olefinic H: δ = 4.30 ppm and 3-H2: δ = 3.24 ppm).

- 141) Abiko A., Moriya O., Filla S. A., Masamune S., Angew. Chem. Int. Ed. Engl., 34, 793–795 (1995).
- 142) Nemoto H., Ma R., Kawamura T., Kamiya M., Shibuya M., J. Org. Chem., 71, 6038–6043 (2006).
- 143) Evans A. C., Longbottom D. A., Matsuoka M., Davis J. E., Turner R., Franckevičius V., Ley S. V., Org. Biomol. Chem., 7, 747–760 (2009).
- 144) Evans A. C., Longbottom D. A., Matsuoka M., Ley S. V., Synlett, 2005, 646–648 (2005).
- 145) Abiko A., Masamune S., Tetrahedron Lett., 37, 1077–1080 (1996).
- 146) An NOE correlation was observed between the olefinic H atom (δ = 4.79 ppm) and 9b-H (δ = 4.62 ppm) in the NOESY spectrum.
- 147) For (-)-33, see: Zhu S.-F., Yu Y.-B., Li S., Wang L.-X., Zhou Q.-L., Angew. Chem. Int. Ed., 51, 8872–8875 (2012).
- 148) Yang S., Zhu S.-F., Zhang C.-M., Song S., Yu Y.-B., Li S., Zhou Q.-L., Tetrahedron, 68, 5172–5178 (2012).
- 149) Zhang Y., Han Z., Li F., Ding K., Zhang A., Chem. Commun. (Camb.), 2010, 156–158 (2010).
- 150) Li S., Zhu S.-F., Zhang C.-M., Song S., Zhou Q.-L., J. Am. Chem. Soc., 130, 8584–8585 (2008).
- 151) For (+)-33, see: Stivala C. E., Zakarian A., J. Am. Chem. Soc., 133, 11936–11939 (2011).
- 152) Miyoshi T., Matsuya S., Tsugawa M., Sato S., Ueda M., Miyata O., Org. Lett., 15, 3374–3377 (2013).
- 153) Miyoshi T., Matsuya S., Tsugawa M., Sato S., Ueda M., Miyata O., Synfacts, 2013, 1109 (2013).
- 154) Kang S. H., Kang S. Y., Lee H.-S., Buglass A. J., Chem. Rev., 105, 4537–4558 (2005).
- 155) Funabashi K., Ratni H., Kanai M., Shibasaki M., J. Am. Chem. Soc., 123, 10784–10785 (2001).
- 156) Sarges R., Howard H. R. Jr., Kelbaugh P. R., J. Org. Chem., 47, 4081–4085 (1982).
- 157) Kleier D. A., Pilgram K. H., J. Heterocycl. Chem., 24, 1643–1647 (1987).
- 158) Saczewski J., Brzozowski Z., Gdaniec M., Tetrahedron, 61, 5303–5309 (2005).
- 159) Sato S., Takeda N., Ueda M., Miyata O., Synthesis (Stuttg.), 48, 882–892 (2016).
- 160) Ghosh A. K., J. Org. Chem., 75, 7967–7989 (2010).
- 161) Nicolaou K. C., Zou B., Dethe D. H., Li D. B., Chen D. Y.-K., Angew. Chem. Int. Ed., 45, 7786–7792 (2006).
- 162) Liu Q., Ferreira E. M., Stoltz B. M., J. Org. Chem., 72, 7352–7358 (2007).
- 163) When PhAlMe2 was used in the sequential rearrangement/phenylation reaction, no methylated product, which would be produced by the addition of a methyl group to the N-(trifluoroacetyl) ketimine, was obtained. In the case of PhAlEt2, a small amount of the ethylated product was detected.
- 164) Stevens C. L., US 3254124 (1966).
- 165) Yokoyama R., Matsumoto S., Nomura S., Higaki T., Yokoyama T., Kiyooka S., Tetrahedron, 65, 5181–5191 (2009).
- 166) Yokoyama T., Yokoyama R., Nomura S., Matsumoto S., Fujiyama R., Kiyooka S., Bull. Chem. Soc. Jpn., 82, 1528–1532 (2009).
- 167) d’Ischia M., Napolitano A., Pezzella A., “Comprehensive Heterocyclic Chemistry III,” ed. by Katritzky A. R., Ramsden C. A., Scriven E. F. V., Taylor R. J. K., Elsevier, Oxford, 2008, p. 353.
- 168) Keay B. A., Hopkins J. M., Dibble P. W., “Comprehensive Heterocyclic Chemistry III,” Katritzky A. R., Ramsden C. A., Scriven, E. F. V., Taylor R. J. K., Elsevier, Oxford, 2008, p. 571.
- 169) Schatz J., Brendgen T., Schühle D., “Comprehensive Heterocyclic Chemistry III,” ed. by Katritzky A. R., Ramsden C. A., Scriven E. F. V., Taylo R. J. K., Elsevier, Oxford, 2008, p. 931.
- 170) Syntheses of Duloxetine hydrochloride are known.
- 171) Träff A., Lihammar R., Bäckvall J.-E., J. Org. Chem., 76, 3917–3921 (2011).
- 172) Suzuki Y., Iwata M., Yazaki R., Kumagai N., Shibasaki M., J. Org. Chem., 77, 4496–4500 (2012).
- 173) Calow A. D. J., Fernández E., Whiting A., Org. Biomol. Chem., 12, 6121–6127 (2014).
- 174) For the synthesis of Tiotropium, see: Banholzer R., Bauer R., Reichl R. E. P., Chem. Abstr., 116, 20937 (1992).
- 175) Wang L., Shen J., Tang Y., Chen Y., Wang W., Cai Z., Du Z., Org. Process Res. Dev., 11, 487–489 (2007).
- 176) van der Meijden M. W., Leeman M., Gelens E., Noorduin W. L., Meekes H., van Enckevort W. J. P., Kaptein B., Vlieg E., Kellogg R. M., Org. Process Res. Dev., 13, 1195–1198 (2009).
- 177) Sadhukhan A., Saravanan S., Khan N. H., Kureshy R. I., Abdi S. H. R., Bajaj H. C., J. Org. Chem., 77, 7076–7080 (2012).
- 178) For the synthesis of Prasugrel hydrochloride, see: Aalla S., Gilla G., Metil D. S., Anumula R. R., Vummenthala P. R., Padi P. R., Org. Process Res. Dev., 16, 240–243 (2012).
- 179) For the synthesis of Rivaroxaban, see: Roehrig S., Straub A., Pohlmann J., Lampe T., Pernerstorfer J., Schlemmer K.-H., Reinemer P., Perzborn E., J. Med. Chem., 48, 5900–5908 (2005).
- 180) For the synthesis of Tiletamine hydrochloride, see: Lapin Y. A., Sanchez I. H., US 6147226 (2000).
- 181) The treatment of Tiletamine with HCl in Et2O (1.0 M) afforded Tiletamine hydrochloride in quantitative yield.
- 182) Takeda N., Furuishi M., Nishijima Y., Futaki E., Ueda M., Shinada T., Miyata O., Org. Biomol. Chem., 16, 8940–8943 (2018).
- 183) Ueda M., Ichimonji A., Nakayama M., Ito S., Takeda N., Yasui M., Chem. Pharm. Bull., 71, 83–92 (2023).
- 184) Sukhorukov A. Y., Adv. Synth. Catal., 362, 724–754 (2020).
- 185) Futaki E., Takeda N., Yasui M., Shinada T., Miyata O., Ueda M., Org. Biomol. Chem., 18, 1563–1566 (2020).
- 186) Takeda N., Suganuma R., Yasui M., Ueda M., Org. Biomol. Chem., 21, 1435–1439 (2023).
- 187) Sukhorukov A. Y., Synlett, 31, 439–449 (2020).