Abstract
Alzheimer’s disease (AD) is the leading cause of senile dementia, and the rapid increase in the frequency of AD cases has been attributed to population aging. However, current drugs have difficulty adequately suppressing symptoms and there is still a medical need for symptomatic agents. On the other hand, it has recently become clear that epigenetic dysfunctions are deeply involved in the development of cognitive impairments. Therefore, epigenetics-related proteins have attracted much attention as drug targets for AD. Early-developed epigenetic inhibitors were inappropriate for AD treatment because of their limited potential for oral administration, blood-brain barrier penetration, high target selectivity, and sufficient dose-limiting toxicity which are essential properties for small molecule drugs targeting chronic neurodegenerative diseases such as AD. In recent years, drug discovery studies have been actively performed to overcome such problems and several novel inhibitors targeting the epigenetics-related proteins are of interest as promising AD therapeutic agents. Here, we review the small molecule inhibitors of histone deacetylase (HDAC), lysine-specific demethylase 1 (LSD1) or bromodomains and extra-terminal domain (BET) protein, that enable memory function improvement in AD model mice.
1. Introduction
Owing to population aging, the incidence of dementia patients is estimated to reach 130 million, worldwide, by 2050.1) Alzheimer’s disease (AD) is one of the most common progressive neurodegenerative diseases which cause dementia in the elderly population and is becoming more prevalent worldwide. Neurodegeneration linked to AD is thought to be induced by an intricate intertwining of neurofibrillary changes in the tau protein neurotoxin, amyloid-beta (Aβ) plaque accumulation in mature neurons, and neuroinflammation that inhibit synaptic function, which plays an important role in the formation and storing of memories.2–5) However, the details regarding the underlying mechanism in the pathology of AD remain unknown, which makes it difficult to establish radical treatment for AD.
As a symptomatic therapy for AD, acetylcholinesterase inhibitors such as donepezil are clinically being used, because they increase the level of acetylcholine in the brain to activate the synaptic transmission.6) However, their effects are temporary, and they are unable to completely stop the progression of the neurological lesions. Several drug discovery studies on targeting tau protein and Aβ pathologies have also been perfomed,7,8) but the developed drugs are still far from clinical stages and being used as curative therapy. In more recent, lecanemab, a humanized immunoglobulin G1 (IgG1) monoclonal antibody that binds with high affinity to Aβ-soluble protofibrils, has been developed by Eisai and Biogen. It reduced Aβ levels in early AD patients and resulted in less decline in cognitive function compared to placebo.9) However, despite these positive outcomes, its efficacy was still insufficient as an 18-month study revealed a cognitive decline reduction rate of only 27%. Thus, the current drug therapy for AD has not sufficiently improved symptoms, suggesting that there is still a medical need for symptomatic agents. Therefore, we need to gain a further understanding of the therapeutic target for the symptoms of AD.
Epigenetics, a mechanism of gene expression that does not involve changes in DNA sequence, has also been reported to be responsible for memory formation and storage in the last two decades.10–12) In particular, enzymatic lysine modifications of histone proteins such as acetylation and methylation are involved in cognitive impairment (Fig. 1). For example, histone deacetylase 2 (HDAC2) regulates synaptic plasticity and long-term changes in neural circuits, negatively regulating learning, and memory.13,14) Other epigenetic proteins including lysine-specific demethylase 1 (LSD1) and bromodomains and extra-terminal domain (BET) protein are also associated with cognitive functions.15,16) Therefore, the epigenetics-related proteins have attracted much attention as a therapeutic target for AD-related symptoms and their inhibition has been reported to improve cognitive impairment,13,17–19) suggesting that small molecule inhibitors targeting the epigenetics-related proteins are expected to act as AD therapeutic agents.
Conventionally, inhibitors of the epigenetics-related proteins have been developed as anti-cancer agents, and some of them have been clinically used or tested; for example, several HDAC inhibitors such as vorinostat and romidepsin are U.S. Food and Drug Administration (FDA)-approved drugs for the treatment of cutaneous T-cell lymphoma20); Tranylcypromine, which is an LSD1 inhibitor and was originally approved by FDA in 1961 for major depressive disorder,21) is currently in clinical trials for acute myelogenous leukemia in combination with all-trans retinoic acid; ZEN003694 (ZEN-3694), a BET inhibitor, is undergoing Phase 2 clinical trials for solid tumors in combination with talazoparib. On the other hand, recently reported epigenetics-related inhibitors are expected to maintain efficacy and minimize dose-limiting toxicity by avoiding substructures that induce side effects or by improving the target selectivity. In addition, some of them can be orally administered and cross the blood-brain barrier. Thus, the novel epigenetics-related inhibitors have attracted much attention as AD therapeutic agents. In this review, we describe the recent advances in small molecule compounds targeting HDACs, LSD1, and BET protein, and discuss their application and potential as therapeutic agents for AD.
2. HDACs and HDAC Inhibitors as AD Therapeutic Agents
HDACs catalyze deacetylation of histone lysine residues and regulate histone acetylation levels.22) The HDAC isoforms are categorized into zinc-dependent and oxidized form of nicotinamide adenine dinucleotide (NAD+)-dependent enzymes: zinc-dependent HDACs, class I (HDACs 1, 2, 3, and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC11): NAD+-dependent HDACs, often called Sirtuin, class III (SIRT1-7). The histone deacetylation by HDACs contributes significantly to the cognitive decline associated with AD-related neurodegeneration.23) Especially, HDAC2, HDAC3, HDAC6, and SIRT2 have been reported to negatively regulate memory formation.13,17,24,25) Therefore, histone acetylation up-regulated by HDAC isoform-selective inhibitors is expected to facilitate learning and memory formation. Furthermore, some small molecule HDAC inhibitors have been reported to improve cognitive impairment in AD model mice.
Vorinostat (Fig. 2), an FDA-approved HDAC inhibitor, was reported to promote memory formation by Tsai and co-workers.13) However, vorinostat has several side effects such as thrombocytopenia, neutropenia, diarrhea, nausea, vomiting, and fatigue,26) because of its poor HDAC isoform-selectivity27) and its hydroxamate structure has raised safety concerns because it might cause potential mutagenicity.28) These side effects are not acceptable for chronic neurodegenerative diseases such as AD. In addition, the brain-to-plasma concentration ratio of vorinostat is estimated to be 4% and its brain permeability is poor.29) In other words, conventional HDAC inhibitors including vorinostat are inappropriate for the treatment of AD, and non-hydroxamate selective inhibitors targeting an HDAC isoform which is related to memory impairment may be preferred for positive and safe therapeutic effects.
BRD4884 and BRD6688 (Fig. 2) exhibited kinetically selective inhibition30) for HDAC2 over HDAC1.31) Both compounds increased lysine acetylation levels of lysine 9 in histone H3 (H3K9) and H4K12 in mouse primary neuronal cell culture assays and restored memory deficits in a cognitive-behavioral model of CK-p25 mice, an animal model representing neurodegenerative disease. Since HDAC1 was reported to provide neuroprotective effects,32) these kinetically HDAC2-selective inhibitors are thought to be promising as AD therapeutic agents. Recently, there has been a movement to understand the structural biology of the kinetically HDAC2-selective inhibition by theoretical analysis. The structural perspective may enable medicinal chemists to design better kinetically selective inhibitors.33)
Rodin-A (Fig. 2), a 2-amino-benzamide HDAC inhibitor, was reported to inhibit the HDAC activity in corepressor of re1-silencing transcription factor (CoREST) complex more strongly than those in Sin3 and NuRD complexes.34) HDAC1/2 are members of the multi-protein co-repressor complexes,35) and especially in the brain, HDAC2 interacts with CoREST and controls neurodegeneration.13) Rodin-A was shown to exert long-term potentiating effects at safe doses on an AD model mouse with five familial Alzheimer’s disease (5xFAD) mutations co-expression and additively increased Aβ production.36) Selective inhibitors for HDACs associated with the CoREST complex, like Rodin-A, may reduce peripheral toxicity in addition to exerting beneficial effects on the nervous system. Thus, CoREST selective HDAC inhibition is a promising therapeutic strategy with fewer side effects.
Compound 17 (Fig. 2), an HDAC2 inhibitor, was obtained by fragment-based ligand discovery.37) Originally, compound 3 was identified as an α-amino-amide zinc binding fragment that bidentately coordinates the catalytic zinc ion of HDACs like hydroxamate or 2-amino-benzamide (Fig. 3a). Subsequent fragment-based optimization, guided by the structural information, disclosed compound 17 (Fig. 3b). Compound 17 showed good plasma and brain exposure when orally administered to mice and was able to induce accumulation of histone acetylation in the brain. Unlike hydroxamate- or 2-aminoanilide-type HDAC inhibitors which are associated with potential toxicity issues,38) non-hydroxamate- or non-2-aminoanilide HDAC inhibitors such as compound 17 are expected to be safer HDAC inhibitors to be used as AD therapeutic agents.
RGFP-966 (Fig. 2) showed a slow-binding profile against HDACs 1–3, with a slight preference for HDAC3.39) RGFP-966 improved spatial learning and memory in a triple transgenic AD mouse model.40) HDAC3 is known to impair spatial memory and causes AD-related plasticity deficits in animal models of AD.17) Another HDAC3-selective inhibitor T24741) (Fig. 2), identified by combinatorial fragment assembly using click chemistry, increased long-term contextual fear memory.42)
PT3 (Fig. 2), a selective HDAC3 inhibitor, exhibited excellent blood–brain barrier permeability and the ability to enhance long-term memory in mouse models of novel object recognition.43) Brain tissue analysis of PT3-treated mice revealed a significant increase in H3K9 acetylation specifically in the hippocampus.
T-518 (Fig. 2), a brain-penetrating HDAC6-selective inhibitor, has oxadiazole moiety and restored impaired novel object recognition on the P301S tau Tg mouse model.44) It was previously reported that overexpression of HDAC6 was observed in the brain of patients with AD,45) and HDAC6 is currently regarded as a therapeutic target for AD. Since HDAC6 regulates acetylation levels of microtubules,46) which are important for axon stability,47) pharmacological HDAC6 inhibition has the potential to improve axon function and is interesting as a treatment strategy for AD.
NCO-141 (Fig. 2) is a selective inhibitor of SIRT2,48) which is abundantly expressed in the brain.49) NCO-141 showed blood-brain barrier penetrating ability and improved spatial learning and memory deficits in the Morris water maze in the five-month-old senescence-accelerated mouse prone 8 (SAMP8) mice.50) Since the SAMP8 is an AD model mouse with age-related cognitive decline51) and SIRT2-mediated epigenetic changes are deeply associated with aging, SIRT2 inhibition is expected to ameliorate age-dependent cognitive impairment and related neuropathology. Accordingly, SIRT2 inhibitors are of interest as agents of neurodegenerative disorders including AD.
As mentioned in this section, remarkable compounds have been developed as HDAC inhibitors that improve cognitive function in AD model mice. Specifically, unique compounds such as isoform-selective HDAC inhibitors, kinetically HDAC2-selective inhibitors, and CoREST-selective HDAC inhibitors have been developed. Moreover, HDAC inhibitors without hydroxamate and 2-amino-benzamide scaffolds which often cause mutagenicity or toxicity have been reported to improve cognitive impairments. It is hoped that further application research on their safety is desirable in the future.
3. LSD1 and LSD1 Inhibitors as AD Therapeutic Agents
LSD1, one of the essential epigenetic regulators, is an oxidative demethylase whose cofactor is flavin adenine dinucleotide (FAD), and its role is to remove methyl groups from histone protein.52,53) Abnormal histone methylation leads to impaired synaptic plasticity and cognitive dysfunction, including neurodegenerative diseases.15) Some studies suggest that LSD1 inhibitors restore cognitive impairments in mice experiments.18,54,55) Thus, small molecule inhibitors which control LSD1 enzymatic activity in the brain would offer a new therapeutic option for the treatment of epigenetic dysregulation-related AD.
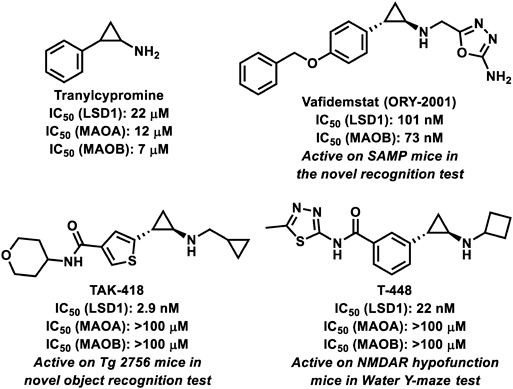
Tranylcypromine (2-phenylcyclopropylamine) (Fig. 4) is the oldest and best-known LSD1 inhibitor, which irreversibly inhibits LSD1 through covalently binding to FAD; FAD first extracts one electron from the nitrogen atom of PCPA to form a cation radical and then, the opening of the cyclopropyl ring occurs followed by covalent bond formation with FAD.56,57) However, since tranylcypromine was originally developed as an inhibitor of monoamine oxygenases (MAOs), which are FAD-dependent oxidases like LSD1, its inhibitory activity and selectivity toward LSD1 were not sufficient.58) In this context, many LSD1 inhibitors have been developed based on tranylcypromine to improve LSD1-ihibitory activity and selectivity. However, many of the irreversible LSD1 inhibitors with sufficient inhibitory activity and selectivity, cause hematologic toxicity such as thrombocytopenia, presumably due to generation of FAD-adducts used to disrupt the interaction between LSD1 and GFI1B.18,59–61) So far, some small molecules that inhibit LSD1 and improve cognitive function, without bringing about hematological side effects, have been identified.
Vafidemstat (ORY-2001) (Fig. 4), a tranylcypromine-based irreversible LSD1 inhibitor, has a brain penetrant property.54) Vafidemstat corrected memory deficits as assessed by a novel object recognition test against SAMP8 mice, a non-transgenic model mouse of accelerated aging and AD. The drug, despite irreversibly inhibiting LSD1, restored memory impairment and behavioral changes at doses that did not trigger hematological side effects. It is the first and only LSD1 inhibitor being developed for AD (Table 1).
Table 1. List of Clinical Trials for Inhibitors of Epigenetic-Related Proteins in the Treatment of AD
| Compound | Study title | NCT number | Status |
|---|
| Vorinostat | Clinical Trial to Determine Tolerable Dosis of Vorinostat in Patients with Mild Alzheimer Disease (VostatAD01) | NCT03056495 | Phase 1 completed |
| RDN-929 | A 28 Day Parallel Group Study to Assess the Effects of RDN-929 | NCT03963973 | Phase 1 terminated |
| RDN-929 | Phase 1 ALKS 1140 in Healthy Adults | NCT05019105 | Phase 1 terminated |
| RDN-929 | Single and Multiple Ascending Dose and Food Effect PK Study in Healthy Adult and Elderly Subjects | NCT03668314 | Phase 1 completed |
| Vafidemstat (ORY-2001) | Testing the Safety and Preliminary Efficacy of the New Drug ORY-2001 in Mild to Moderate Alzheimer’s Disease (ETHERAL-U.S.) | NCT03867253 | Phase 2 completed |
| RVX-208 | Reference 76 | | |
TAK-418 (Fig. 4), a tranylcypromine-based irreversible and highly selective inhibitor of LSD1, was reported to be effective on Tg2576 mice which is a mouse model for memory impairment due to aging and overexpression of AD-related mutant amyloid precursor protein (APP).18) TAK-418, despite being an irreversible LSD1 inhibitor, consistently ameliorated social deficits in animal models of neurodevelopmental disorders without dissociating the LSD1-GFI1B complex. The clinical applications of this drug as AD treatment are expected.
T-448 (Fig. 4), an analog of TAK-418, partially restored learning function in mice with elevated H3K4 methylation levels in the brain and impaired N-methyl-D-aspartate (NMDA) receptor function in a capacity that ensured hematological safety.55)
The tranylcypromine derivatives have been developed as LSD1 inhibitors that do not show hematological toxicity derived from inhibition of the interaction between LSD1 and GFI1B, and these have been reported to improve cognitive function in AD model mice. Furthermore, several brain-penetrating LSD1 inhibitors have been disclosed; e.g., DDP3800362) and NCD38,63,64) which were originally identified as anticancer agents, are highly capable of crossing the blood–brain barrier. These inhibitors are expected to be applied to AD therapy and these findings help with the further development of novel LSD1 inhibitors which potentially improve cognitive impairment.
4. BET Proteins and BET Inhibitors as AD Therapeutic Agents
BET proteins are known as epigenetic “readers” that recognize acetylated lysine residues on histone tails.65) There are four BET proteins in humans, namely, BRD2, BRD3, BRD4, and testis-specific BRDT. BET proteins are highly specific for acetylated histone tails and have the highest affinity for the acetyl group of H4K12 (H4K12Ac) and H4K5Ac, and BRD2 and BRD4 preferentially bind to H4K12Ac.66,67) Furthermore, H4K5Ac and H4K12Ac have been found to be specifically associated with memory function and age-related memory impairment.13,68) Therefore, targeting the histone acetylation has been regarded as a novel approach for the treatment of dementia. Thus, the development of small molecules that modulate BET protein function is an interesting approach for the treatment of late-stage cognitive disorders such as AD.
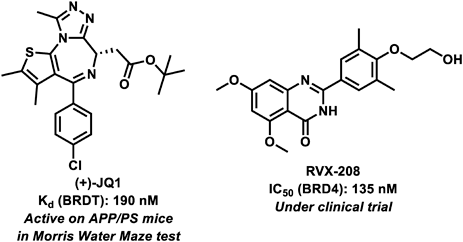
JQ1 (Fig. 5), a small molecule inhibitor of BRD2, BRD3, BRD4, and BRDT, has brain-penetrating ability.69) JQ1 improved cognitive performance and long-term potentiation in wild-type and APP mouse model of AD.19) Furthermore, JQ1 suppressed inflammation and tau phosphorylation at Ser396 in the brain of the 3xTg AD model mouse,70) which represents an early event in AD.71) However, seven-month-old 3xTg mice treated with JQ1 showed no cognitive function improvement in behavioral tests. This may be due to the relatively young age of the cohort of treated mice and the late onset of behavioral deficits. 3xTg mice at this age begin to develop deficits in spatial working memory,72) while more severe deficits in recognition and reference memory appear only between the ages of nine and 12 months.73) Further validation of experiments using JQ1 for long periods of time and in different age frames is needed to determine the effects of JQ1 on cognition in mice.
RVX-208 (Fig. 5) is a BET inhibitor74) and was clinically tested for its effect on cognitive function by collecting results from the Montreal Cognitive Assessment (MoCA), a simple screening test for cognitive impairment,75) in participants aged 70 years and older.76) In this clinical trial, RVX-208 was associated with improved cognitive function. As mentioned above, BET protein inhibitors warrant further studies on cognitive impairment in the late elderly population.
As mentioned above, several BET inhibitors are known to improve cognitive function in AD model mice or a late-aged population in clinical trials, suggesting that BET proteins have potential as therapeutic targets for AD. However, research on the application of BET protein inhibitors to AD is still limited. It is expected that more BET inhibitors targeting the improvement of cognitive function will be developed in the future.
5. Conclusion and Outlook
The number of patients with AD has been increasing rapidly in line with population aging, while fundamental preventive or curative treatment methods are lacking. The process of AD is accompanied by the abnormal deposition of two proteins, Aβ and tau. Drugs targeting these pathologies have been developed; however, they cannot stop the progression of AD, and AD patients continue to suffer its symptoms. Thus, therapeutic strategies targeting the mechanisms causing the symptoms, like inhibition of epigenetics-related proteins, have been required. As mentioned in this review, inhibitors of the epigenetics-related proteins such as HDAC, LSD1, and BET proteins are in focus as novel potential therapeutic agents for AD. Indeed, some of the small molecule compounds presented here have shown a trend toward improving dementia in animal models and clinical trials alike.
Table 1 provides a list of epigenetics-related protein inhibitors whose clinical trials have been underway for the treatment of AD. Vorinostat, a pan-HDAC inhibitor, was tested in patients with mild AD, and its safety study was completed. Additionally, three phase 1 studies of RDN-929, an analog of RodinA, have been conducted. One study (NCT03668314) was completed, while the others (NCT05019105 and NCT03963973) were terminated. Vafidemstat (ORY-2001), an LSD1 inhibitor, demonstrated a reduction in inflammatory and neuropathic biomarkers in Phase 2 clinical trials. However, it did not show improvement in cognitive signals.
Despite the development of potent inhibitors, there have been instances of insufficient safety or efficacy in clinical trials for AD. In other words, for the development of drugs targeting the central nervous system such as AD drugs, it is essential to equip them with both brain permeability and safety. In general, brain-permeable drugs have the following features; lower molecular weight, lower Clog P, moderate topological polar surface area, higher ClogD, lower hydrogen-bond donors, and lower pKa values.77) In addition, there should be a need to avoid a pharmacophore causing toxicity and mutagenesis to develop safe drugs; non-hydroxamate or non-2-aminobenzamide may be favorable in the case of HDAC inhibitors. Identification of the drugs with not only enough efficacy but also these features should lead to discovering successful outcomes in clinical trials.
In addition to the epigenetics-related enzymes mentioned in this review, other enzymes have also been reported to be associated with AD progression. For instance, it was reported that inhibition of G9a/GLP reduced demethylation of H3K9 levels in the hippocampal region and rescues cognitive deficits in AD model mice.78,79) Moreover, an increase in levels of lysine methyltransferase (KMT) was observed in the prefrontal cortex of patients with AD and postmortem tissue of P301S tau mice, and inhibition of KMTs ameliorated cognitive deficits in these mice.80) It would be interesting to develop KMT inhibitors with sufficient brain penetration ability and selectivity for clinical drug development.
As described in this review, inhibition of the epigenetics-related proteins is promising as a new strategy for AD therapy. Because the symptomatic agents for AD are in high demand, the epigenetics-related proteins are interesting therapeutic targets of such agents. Especially, some of the inhibitors described in this review are effective in the improvement of cognitive function not only in mouse models of AD but also in the clinical trial stage. In addition, epigenetic-related enzyme inhibitors may be interesting as therapeutic agents for other types of dementia such as frontotemporal dementia (FTD).81) Indeed, vorinostat has been suggested to enhance the expression of progranulin whose levels in patients suffering from FTD are often reduced by GRN mutations resulting in impaired transcription. Thus, HDAC inhibitors hold promise as novel therapeutic agents for neurodegenerative disorders such as FTD. We hope that such drugs will provide dementia patients and their families with a higher QOL in the future.
Acknowledgments
This work was supported by Grants for JSPS KAKENHI, “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from MEXT and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research; BINDS) from the Japan Agency for Medical Research and Development (AMED) (22ama121041j0001).
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Frisoni G. B., Altomare D., Thal D. R., Ribaldi F., van der Kant R., Ossenkoppele R., Blennow K., Cummings J., van Duijn C., Nilsson P. M., Dietrich P.-Y., Scheltens P., Dubois B., Nat. Rev. Neurosci., 23, 53–66 (2022).
- 2) Ogunmokun G., Dewanjee S., Chakraborty P., Valupadas C., Chaudhary A., Kolli V., Anand U., Vallamkondu J., Goel P., Paluru H. P. R., Gill K. D., Reddy P. H., Feo V. D., Kandimalla R., Cells, 10, 2790 (2021).
- 3) Katsuse O., Lin W.-L., Lewis J., Hutton M. L., Dickson D. W., Neurosci. Lett., 409, 95–99 (2006).
- 4) Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J., Nature (London), 416, 535–539 (2002).
- 5) Roy E. R., Wang B., Wan Y.-W., Chiu G., Cole A., Yin Z., Propson N. E., Xu Y., Jankowsky J. L., Liu Z., Lee V. M.-Y., Trojanowski J. Q., Ginsberg S. D., Butovsky O., Zheng H., Cao W., J. Clin. Invest., 130, 1912–1930 (2020).
- 6) Marucci G., Buccioni M., Ben D. D., Lambertucci C., Volpini R., Amenta F., Neuropharmacology, 190, 108352 (2021).
- 7) Congdon E. E., Sigurdsson E. M., Nat. Rev. Neurol., 14, 399–415 (2018).
- 8) Panza F., Lozupone M., Logroscino G., Imbimbo B. P., Nat. Rev. Neurol., 15, 73–88 (2019).
- 9) van Dyck C. H., Swanson C. J., Aisen P., Bateman R. J., Chen C., Gee M., Kanekiyo M., Li D., Reyderman L., Cohen S., Froelich L., Katayama S., Sabbagh M., Vellas B., Watson D., Dhadda S., Irizarry M., Kramer L. D., Iwatsubo T., N. Engl. J. Med., 388, 9–21 (2023).
- 10) Guan Z., Giustetto M., Lomvardas S., Kim J.-H., Miniaci M. C., Schwartz J. H., Thanos D., Kandel E. R., Cell, 111, 483–493 (2002).
- 11) Alarcó J. M., Malleret G. L., Touzani K., Vronskaya S., Ishii S., Kandel E. R., Barco A., Neuron, 42, 947–959 (2004).
- 12) Korzus E., Rosenfeld M. G., Mayford M., Neuron, 42, 961–972 (2004).
- 13) Guan J.-S., Haggarty S. J., Giacometti E., Dannenberg J.-H., Joseph N., Gao J., Nieland T. J. F., Zhou Y., Wang X., Mazitschek R., Bradner J. E., DePinho R. A., Jaenisch R., Tsai L.-H., Nature (London), 459, 55–60 (2009).
- 14) Gräff J., Rei D., Guan J.-S., Wang W.-Y., Seo J., Hennig K. M., Nieland T. J. F., Fass D. M., Kao P. F., Kahn M., Su S. C., Samiei A., Joseph N., Haggarty S. J., Delalle I., Tsai L.-H., Nature (London), 483, 222–226 (2012).
- 15) Basavarajappa B. S., Subbanna S., Int. J. Mol. Sci., 22, 4654 (2021).
- 16) Nikkar R., Esmaeili-bandboni A., Badrikoohi M., Babaei P., Metab. Brain Dis., 37, 1119–1131 (2022).
- 17) Zhu X., Wang S., Yu L., Jin J., Ye X., Liu Y., Xu Y., Aging Cell, 16, 1073–1082 (2017).
- 18) Baba R., Matsuda S., Maeda R., Murakami K., Yamamoto Y., Nakatani A., Kimura H., ACS Chem. Neurosci., 13, 313–321 (2022).
- 19) Benito E., Ramachandran B., Schroeder H., Schmidt G., Urbanke H., Burkhardt S., Capece V., Dean C., Fischer A., Transl. Psychiatry, 7, e1239 (2017).
- 20) Bondarev A. D., Attwood M. M., Jonsson J., Chubarev V. N., Tarasov V. V., Schiöth H. B., Br. J. Clin. Pharmacol., 87, 4577–4597 (2021).
- 21) Noce B., Bello E. D., Fioravanti R., Mai A., Front. Pharmacol., 14, 1120911 (2023).
- 22) Glozak M. A., Sengupta N., Zhang X., Seto E., Gene, 363, 15–23 (2005).
- 23) Levenson J. M., O’Riordan K. J., Brown K. D., Trinh M. A., Molfese D. L., Sweatt J. D., J. Biol. Chem., 279, 40545–40559 (2004).
- 24) Govindarajan N., Rao P., Burkhardt S., Sananbenesi F., Schlüter O. M., Bradke F., Lu J., Fischer A., EMBO Mol. Med., 5, 52–63 (2013).
- 25) Silva D. F., Esteves A. R., Oliveira C. R., Cardoso S. M., Mol. Neurobiol., 54, 4021–4040 (2017).
- 26) Gryder B. E., Sodji Q. H., Oyelere A. K., Future Med. Chem., 4, 505–524 (2012).
- 27) Ho T. C. S., Chan A. H. Y., Ganesan A., J. Med. Chem., 63, 12460–12484 (2020).
- 28) Wang C. Y., Lee L. H., Antimicrob. Agents Chemother., 11, 753–755 (1977).
- 29) Kim M., Kizibash S. H., Laramy J. K., Gampa G., Parrish K. E., Sarkaria J. N., Elmquist W. F., Pharm. Res., 35, 177 (2018).
- 30) Copeland R. A., Pompliano D. L., Meek T. D., Nat. Rev. Drug Discov., 5, 730–739 (2006).
- 31) Wagner F. F., Zhang Y.-L., Fass D. M., et al., Chem. Sci. (Camb.), 6, 804–815 (2015).
- 32) Kim D., Frank C. L., Dobbin M. M., Tsunemoto R. K., Tu W., Peng P. L., Guan J.-S., Lee B.-H., Moy L. Y., Giusti P., Broodie N., Mazitschek R., Delalle I., Haggarty S. J., Neve R. L., Lu Y., Tsai L.-H., Neuron, 60, 803–817 (2008).
- 33) Mishima H., Itoh Y., Kurohara T., Suzuki T., Asada N., Kusakabe K., Okamoto Y., J. Comput. Chem., 44, 1604–1609 (2023).
- 34) Fuller N. O., Pirone A., Lynch B. A., Hewitt M. C., Quinton M. S., McKee T. D., Ivarsson M., ACS Chem. Neurosci., 10, 1729–1743 (2019).
- 35) Itoh Y., Takada Y., Yamashita Y., Suzuki T., Curr. Opin. Chem. Biol., 67, 102130 (2022).
- 36) Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Eldik L. V., Berry R., Vassar R., J. Neurosci., 26, 10129–10140 (2006).
- 37) Tamanini E., Miyamura S., Buck I. M., et al., ACS Med. Chem. Lett., 13, 1591–1597 (2022).
- 38) Mottamal M., Zheng S., Huang T. L., Wang G., Molecules, 20, 3898–3941 (2015).
- 39) Moreno-Yruela C., Olsen C. A., ACS Med. Chem. Lett., 13, 779–785 (2022).
- 40) Janczura K. J., Volmar C.-H., Sartor G. C., Rao S. J., Ricciardi N. R., Lambert G., Brothers S. P., Wahlestedt C., Proc. Natl. Acad. Sci. U.S.A., 115, E11148–E11157 (2018).
- 41) Suzuki T., Kasuya Y., Itoh Y., Ota Y., Zhan P., Asamitsu K., Nakagawa H., Okamoto T., Miyata N., PLOS ONE, 8, e68669 (2013).
- 42) Uchida S., Teubner B. J. W., Hevi C., Hara K., Kobayashi A., Dave R. M., Shintaku T., Jaikhan P., Yamagata H., Suzuki T., Watanabe Y., Zakharenko S. S., Shumyatsky G. P., Cell Rep., 18, 352–366 (2017).
- 43) Pulya S., Mahale A., Bobde Y., Routholla G., Patel T., Swati, Biswas S., Sharma V., Kulkarni O. P., Ghosh B., ACS Chem. Neurosci., 12, 883–892 (2021).
- 44) Onishi T., Maeda R., Terada M., Sato S., Fujii T., Ito M., Hashikami K., Kawamoto T., Tanaka M., Sci. Rep., 11, 15423 (2021).
- 45) Ding H., Dolan P. J., Johnson G. V. W., J. Neurochem., 106, 2119–2130 (2008).
- 46) Brindisi M., Saraswati A. P., Brogi S., Gemma S., Butini S., Campiani G., J. Med. Chem., 63, 23–39 (2020).
- 47) Portran D., Schaedel L., Xu Z., Théry M., Nachury M. V., Nat. Cell Biol., 19, 391–398 (2017).
- 48) Suzuki T., Khan M. N. A., Sawada H., Imai E., Itoh Y., Yamatsuta K., Tokuda N., Takeuchi J., Seko T., Nakagawa H., Miyata N., J. Med. Chem., 55, 5760–5773 (2012).
- 49) Jayasena T., Poljak A., Braidy N., Zhong L., Rowlands B., Muenchhoff J., Grant R., Smythe G., Teo C., Raftery M., Sachdev P., Sci. Rep., 6, 35391 (2016).
- 50) Diaz-Perdigon T., Belloch F. B., Ricobaraza A., Elboray E. E., Suzuki T., Tordera R. M., Puerta E., Neuropsychopharmacology, 45, 347–357 (2020).
- 51) Tomobe K., Nomura Y., Neurochem. Res., 34, 660–669 (2009).
- 52) Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y., Cell, 119, 941–953 (2004).
- 53) Karasulu B., Patil M., Thiel W., J. Am. Chem. Soc., 135, 13400–13413 (2013).
- 54) Maes T., Mascaró C., Rotllant D., Lufino M. M. P., Estiarte A., Guibourt N., Cavalcanti F., Griñan-Ferré C., Pallàs M., Nadal R., Armario A., Ferrer I., Ortega A., Valls N., Fyfe M., Martinell M., Palomino J. C. C., Arjol C. B., PLOS ONE, 15, e0233468 (2020).
- 55) Matsuda S., Baba R., Oki H., Morimoto S., Toyofuku M., Igaki S., Kamada Y., Iwasaki S., Matsumiya K., Hibino R., Kamada H., Hirakawa T., Iwatani M., Tsuchida K., Hara R., Ito M., Kimura H., Neuropsychopharmacology, 44, 1505–1512 (2019).
- 56) Dong J., Pervaiz W., Tayyab B., Li D., Kang L., Zhang H., Gong H., Ma X., Li J., Agboyibor C., Bi Y., Liu H., Eur. J. Med. Chem., 240, 114564 (2022).
- 57) Schmidt D. M. Z., McCafferty D. G., Biochemistry, 46, 4408–4416 (2007).
- 58) Ji Y.-Y., Lin S.-D., Wang Y.-J., Su M.-B., Zhang W., Gunosewoyo H., Yang F., Li J., Tang J., Zhou Y.-B., Yu L.-F., Eur. J. Med. Chem., 141, 101–112 (2017).
- 59) Ishikawa Y., Gamo K., Yabuki M., et al., Mol. Cancer Ther., 16, 273–284 (2017).
- 60) Yamamoto R., Kawahara M., Ito S., Satoh J., Tatsumi G., Hishizawa M., Suzuki T., Andoh A., Oncotarget, 9, 21007–21021 (2018).
- 61) Hattori Y., Matsumoto S., Morimoto S., Daini M., Toyofuku M., Matsuda S., Baba R., Murakami K., Iwatani M., Oki H., Iwasaki S., Matsumiya K., Tominari Y., Kimura H., Ito M., Eur. J. Med. Chem., 239, 114522 (2022).
- 62) Vianello P., Botrugno O. A., Cappa A., Zuffo R. D., Dessanti P., Mai A., Marrocco B., Mattevi A., Meroni G., Minucci S., Stazi G., Thaler F., Trifiró P., Valente S., Villa M., Varasi M., Mercurio C., J. Med. Chem., 59, 1501–1517 (2016).
- 63) Ogasawara D., Itoh Y., Tsumoto H., Kakizawa T., Mino K., Fukuhara K., Nakagawa H., Hasegawa M., Sasaki R., Mizukami T., Miyata N., Suzuki T., Angew. Chem. Int. Ed., 52, 8620–8624 (2013).
- 64) Alejo S., Palacios B. E., Venkata P. P., et al., Neuro-oncol., 25, 1249–1261 (2023).
- 65) Belkina A. C., Denis G. V., Nat. Rev. Cancer, 12, 465–477 (2012).
- 66) LeRoy G., Chepelev I., DiMaggio P. A., Blanco M. A., Zee B. M., Zhao K., Garcia B. A., Genome Biol., 13, R68 (2012).
- 67) Umehara T., Nakamura Y., Jang M. K., Nakano K., Tanaka A., Ozato K., Padmanabhan B., Yokoyama S., J. Biol. Chem., 285, 7610–7618 (2010).
- 68) Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R. C., Cota P., Wittnam J. L., Gogol-Doering A., Opitz L., Salinas-Riester G., Dettenhofer M., Kang H., Farinelli L., Chen W., Fischer A., Science, 328, 753–756 (2010).
- 69) Matzuk M. M., McKeown M. R., Filippakopoulos P., Li Q., Ma L., Agno J. E., Lemieux M. E., Picaud S., Yu R. N., Qi J., Knapp S., Bradner J. E., Cell, 150, 673–684 (2012).
- 70) Magistri M., Velmeshev D., Makhmutova M., Patel P., Sartor G. C., Volmar C.-H., Wahlestedt C., Faghihi M. A., Curr. Alzheimer Res., 13, 985–995 (2016).
- 71) Mondragon-Rodriguez S., Perry G., Luna-Munoz J., Acevedo-Aquino M. C., Williams S., Neuropathol. Appl. Neurobiol., 40, 121–135 (2014).
- 72) Clinton L. K., Billings L. M., Green K. N., Caccamo A., Ngo J., Oddo S., McGaugh J. L., LaFerla F. M., Neurobiol. Dis., 28, 76–82 (2007).
- 73) Filali M., Lalonde R., Theriault P., Julien C., Calon F., Planel E., Behav. Brain Res., 234, 334–342 (2012).
- 74) Picaud S., Wells C., Felletar I., Brotherton D., Martin S., Savitsky P., Diez-Dacal B., Philpott M., Bountra C., Lingard H., Fedorov O., Müller S., Brennan P. E., Knapp S., Filippakopoulos P., Proc. Natl. Acad. Sci. U.S.A., 110, 19754–19759 (2013).
- 75) Nasreddine Z. S., Phillips N. A., Bédirian V., Charbonneau S., Whitehead V., Collin I., Cummings J. L., Chertkow H., J. Am. Geriatr. Soc., 53, 695–699 (2005).
- 76) Cummings J., Schwartz G. G., Nicholls S. J., Khan A., Halliday C., Toth P. P., Sweeney M., Johansson J. O., Wong N. C. W., Kulikowski E., Kalantar-Zadeh K., Lebioda K., Ginsberg H. N., Winblad B., Zetterberg H., Ray K. K., J. Alzheimers Dis., 83, 1703–1715 (2021).
- 77) Wager T. T., Hou X., Verhoest P. R., Villalobos A., ACS Chem. Neurosci., 7, 767–775 (2016).
- 78) Zheng Y., Liu A., Wang Z.-J., Cao Q., Wang W., Lin L., Ma K., Zhang F., Wei J., Matas E., Cheng J., Chen G.-J., Wang X., Yan Z., Brain, 142, 787–807 (2019).
- 79) Griñán-Ferré C., Marsal-García L., Bellver-Sanchis A., Kondengaden S. M., Turga R. C., Vázquez S., Pallàs M., Aging (Albany NY), 11, 11591–11608 (2019).
- 80) Cao Q., Wang W., Williams J. B., Yang F., Wang Z.-J., Yan Z., Sci. Adv., 6, eabc8096 (2020).
- 81) She A., Kurtser I., Reis S. A., Hennig K., Lai J., Lang A., Zhao W.-N., Mazitschek R., Dickerson B. C., Herz J., Haggarty S. J., Cell Chem. Biol., 24, 892–906 (2017).