Regular Article
Generation and Coupling of Radical Species from α-Alkoxy Bridgehead Carboxylic Acid, Selenide, Telluride, Acyl Selenide, and Acyl Telluride
2024 Volume 72 Issue 8 Pages 767-771
Details
2024 Volume 72 Issue 8 Pages 767-771
α-Alkoxy bridgehead radicals enable intermolecular construction of sterically congested C–C bonds due to their sterically accessible nature. We implemented these radical species into total syntheses of various densely oxygenated natural products and demonstrated their exceptional versatility. Herein, we employed different precursors to generate the same α-alkoxy bridgehead radical and compared the efficacy of the precursors for coupling reactions. Specifically, the bridgehead radical of the trioxaadamantane structure was formed from α-alkoxy carboxylic acid, selenide/telluride, and acyl selenide/acyl telluride, and reacted with 4-((tert-butyldimethylsilyl)oxy)cyclopent-2-en-1-one and 5-oxo-1-cyclopentene-1-carbonitrile. The efficiency of the bridgehead radical formation and subsequent coupling reaction significantly depended on the structures of the precursors and acceptors as well as the reaction conditions. Our findings provide new insights for selecting the appropriate substrates of key coupling reactions in the total synthesis of complex natural products.
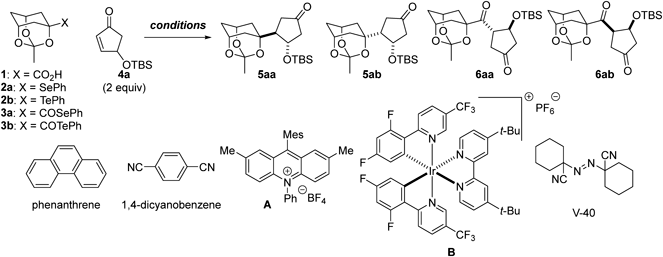
Bridgehead radicals of cage structures differ from common radicals in their structures and reactivities.1) As the β-carbons of bridgehead positions are tied back, steric congestion around the radicals is minimized and stereochemical inversion of the bridgehead radicals is inhibited. Accordingly, the bridgehead radicals allow for the construction of sterically hindered C–C bonds in a stereospecific manner. We are interested in exploiting these distinctive characteristics for natural product synthesis and have explored the possibilities for implementing bridgehead radicals in synthetic routes to complex natural products.2–4) Over the years, we have demonstrated the exceptional versatility of α-alkoxy bridgehead radicals in building polycyclic architectures with multiple oxygen functionalities. We reported that α-alkoxy selenide and cyclopentenone functioned as the precursor and acceptor of the nucleophilic bridgehead radical, respectively, for seven-membered ring cyclization in the total synthesis of crotophorbolone in 20155) and in the unified total syntheses of crotophorbolone, prostratin, phorbol, and resiniferatoxin in 2021.6) In the total synthesis of resiniferatoxin in 2022,7) the seven-membered ring was formed from α-alkoxy carboxylic acid and cyclopentenone. On the other hand, an intermolecular three-component reaction of α-alkoxy selenide, 4-((tert-butyldimethylsilyl)oxy)cyclopent-2-en-1-one (4-TBSoxy-cyclopent-2-en-1-one) (4a), and allylstannane was demonstrated in the total synthesis of resiniferatoxin in 2017,8) while an intermolecular two-component reaction between α-alkoxyacyl telluride and alkylidenemalononitrile was used as the key transformation in the total synthesis of batrachotoxin in 2023.9)
In these achievements, we employed a variety of bridgehead radical precursors and acceptors for the stereospecific inter- and intramolecular reactions. To enhance the synthetic utility of the methodology, we aimed to gain deeper insights into the radical-forming propensity of the precursors and the radical coupling efficiency of the acceptors. Here, we investigated the generation of the α-alkoxy bridgehead radical of the trioxaadamantane structure from carboxylic acid, selenide/telluride, and acyl selenide/acyl telluride, and subsequent coupling of the nucleophilic radical with 4-TBSoxy-cyclopent-2-en-1-one and 5-oxo-1-cyclopentene-1-carbonitrile. These comprehensive comparison data uncovered the distinct roles of the radical generating and accepting functionalities and provide a new design principle for substrates in radical-based natural product synthesis.
In this study, we planned to utilize five different radical precursors including 3-methyl-2,4,10-trioxaadamantane-1-carboxylic acid (1)10) (Chart 1). Four precursors, selenide/telluride 2a/2b and acyl selenide/acyl telluride 3a/3b, were readily derivatized from 1. Carboxylic acid 1 was converted to the corresponding Barton ester,11–13) which was irradiated by an Hg lamp in the presence of (PhSe)2 and (PhTe)2 to produce selenide 2a (71% yield) and telluride 2b (83% yield), respectively.10,14,15) Treatment of 1 with n-Bu3P and (PhSe)2 afforded acyl selenide 3a in 85% yield, while acyl telluride 3b was obtained in 78% yield via formation of the activated ester with i-BuOCOCl from 1 and subsequent reaction with (PhTe)2 and NaBH4.

a) EDCI = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, NMM = N-methylmorpholine.
The thus-prepared five radical precursors were subjected to coupling reactions with 4-TBSoxy-cyclopent-2-en-1-one (4a) (Table 1). First, four distinct photo-irradiation conditions were applied to carboxylic acid 1 and 4a (2 equivalents (equiv.)).16–19) When K2S2O8 (3 equiv.) and K2HPO4 (3 equiv.) were utilized as the oxidant and base, respectively, in MeCN/H2O/dimethyl sulfoxide (DMSO) at 70 °C (entry 1),20) adduct 5aa formed in only 8.2% yield and 1 was recovered in 18% yield, indicating the slow oxidation of the in situ-generated carboxylate by K2S2O8. Treatment of 1 and 4a with phenanthrene and 1,4-dicyanobenzene in the presence of NaOH with Hg lamp irradiation21,22) resulted in decomposition (entry 2), presumably due to β-elimination of the TBSoxy group of 4a by the strong base. In contrast, the photoredox reaction using acridinium catalyst A (10 mol%),23,24) K2HPO4, and blue LED light in MeCN effectively transformed 1 and 4a into adduct 5aa (49%) and its epimer 5ab (2.0%) in a stereoselective manner (5aa/5ab = 25 : 1, entry 3). The reagent combination of {Ir[dF(CF3)ppy]2(dtbbpy)}PF6 (B, 10 mol%) and K2HPO4 (1.5 equiv.) in N,N-dimethylformamide (DMF) under blue LED irradiation25) improved the yields of adducts 5aa and 5ab (5aa: 63%, 5ab: 3.8%, 5aa/5ab = 17 : 1, entry 4). The selective formation of 5aa over its epimer 5ab is attributable to the preferential approach of the radical from the opposite face of the preexisting TBSoxy group. The structure of 5aa was previously determined,10) and that of 5ab was established by X-ray crystallographic analysis (Chart 2).
 | |||||||
|---|---|---|---|---|---|---|---|
| Entrya) | SM | X | Conditions | Yield [%]b) | |||
| 5aa | 5ab | 6aa | 6ab | ||||
| 1 | 1 | CO2H | K2S2O8 (3 equiv.), K2HPO4 (3 equiv.) | 8.2 | 0 | 0 | 0 |
| MeCN/H2O/DMSO (4/1/1, 0.05 M), 70 °C | |||||||
| 2 | 1 | CO2H | Phenanthrene (1 equiv.) | 0 | 0 | 0 | 0 |
| 1,4-Dicyanobenzene (1 equiv.) | |||||||
| NaOH (1 equiv.), MeCN/H2O (9/1, 0.02 M) | |||||||
| Hg lamp, r.t. | |||||||
| 3c) | 1 | CO2H | A (10 mol%), K2HPO4 (1.2 equiv.) | 49 | 2.0 | 0 | 0 |
| MeCN (0.04 M), blue LED, 38 °C | |||||||
| 4 | 1 | CO2H | B (10 mol%), K2HPO4 (1.5 equiv.) | 63 | 3.8 | 0 | 0 |
| DMF (0.1 M), blue LED, 38 °C | |||||||
| 510) | 2a | SePh | n-Bu3SnH (6 equiv.), V-40 (60 mol%) | 62 | 3.8 | 0 | 0 |
| toluene (0.02 M), reflux | |||||||
| 615) | 2b | TePh | Et3B (3 equiv.), air, CH2Cl2 (0.1 M), 0 °C | 72d) | 0 | 0 | 0 |
| 7 | 3a | COSePh | n-Bu3SnH (2 equiv.), V-40 (35 mol%) | 26d) | 3.2 | 0 | 0 |
| toluene (0.02 M), reflux | |||||||
| 8e) | 3b | COTePh | Et3B (3 equiv.), air | 29d) | 1.7 | 6.6 | 3.4 |
| CH2Cl2 (0.02 M), r.t. | |||||||
a) 6–43 mg scale. b) Yield calculated from 1H-NMR using CH2Br2 as an internal standard. c) 3 equiv. of 4a was used. d) Isolated yield. TBS = tert-butyldimethylsilyl, V-40 = 1,1′-azobis(cyclohexane-1-carbonitrile). e) At the 0.1 M concentration, 5aa, 6aa, and 6ab were obtained in 17, 16, and 18% 1H-NMR yields, respectively.

We previously optimized the coupling reactions of α-alkoxy selenide 2a and α-alkoxy telluride 2b with 4a10,15,26) (Table 1). Selenide 2a and 4a were converted into adducts 5aa (62%) and 5ab (3.8%) using n-Bu3SnH (6 equiv.) and radical initiator V-40 (60 mol%) in refluxing toluene (5aa/5ab = 16 : 1, entry 5).27) Telluride 2b and 4a underwent the coupling reaction at 0 °C in the presence of Et3B (3 equiv.) and air in CH2Cl2,28) giving rise to 5aa as a sole isolable isomer in 72% yield (entry 6).29,30) It is noteworthy that the conditions in entry 6 failed to generate the radical from selenide 2a (97% recovery of 2a), likely due to the stronger C–Se bond (bond dissociation energy: 59.8 kcal/mol) compared with the C–Te bond (52.4 kcal/mol).31)
Next, α-alkoxyacyl selenide 3a and α-alkoxyacyl telluride 3b were applied as radical precursors (Table 1). Heating acyl selenide 3a and 4a with n-Bu3SnH (2 equiv.) and radical initiator V-40 (35 mol%) in toluene at 110 °C furnished diastereomers 5aa and 5ab in 26 and 3.2% yields, respectively (5aa/5ab = 8.1 : 1, entry 7). Alternatively, acyl telluride 3b and 4a were treated with Et3B (3 equiv.) and air in CH2Cl2 at room temperature, providing adducts 5aa (29%) and 5ab (1.7%) (5aa/5ab = 17 : 1, entry 8). In this reaction, 6aa (6.6%) and its epimer 6ab (3.4%) were also obtained via direct addition of the acyl radical intermediate to 4a. Partial formation of 6aa/6ab in addition to 5aa/5ab showed slow decarbonylation at room temperature (entry 8) compared with that at 110 °C (entry 7). The newly generated 6aa and 6ab were structurally determined by X-ray crystallographic analyses (Chart 2).
To further investigate the disparate characters of the five radical precursors, 1, 2a/2b, and 3a/3b, the radical acceptor was changed from 4-TBSoxy-cyclopent-2-en-1-one (4a) to 5-oxo-1-cyclopentene-1-carbonitrile (4b)32) (Table 2). Because of the additional electron-withdrawing cyanide, 4b possesses lower lowest unoccupied molecular orbital (LUMO) energy than 4a and exhibits higher reactivity toward nucleophilic bridgehead radicals. The coupling reaction did not proceed, however, when using carboxylic acid 1 and photoredox catalyst B under the photo-irradiation conditions (entry 1). Only the starting material 1 was recovered in 32% yield. The contrasting results between entry 4 in Table 1 and entry 1 in Table 2 likely originate from the base instability of 4b: the γ-position of the cyanide-activated enone would be readily deprotonated by K2HPO4, and the resultant anionic species would undergo unproductive pathways. In fact, treatment of 4b with K2HPO4 in MeCN at room temperature without B or blue LED light merely generated unidentified polar compounds, indicating the short lifetime of 4b under basic conditions.
 | ||||||
|---|---|---|---|---|---|---|
| Entrya) | SM | X | Conditions | 2-Step yield [%]b) | ||
| 7 | 8 | 1 | ||||
| 1 | 1 | CO2H | B (10 mol%), K2HPO4 (1.5 equiv.) | 0 | 0 | 32 |
| DMF (0.1 M), blue LED, 38 °C | ||||||
| 2 | 2a | SePh | n-Bu3SnH (2 equiv.), V-40 (40 mol%) | 58c) | 0 | 0 |
| toluene (0.05 M), reflux | ||||||
| 3 | 2b | TePh | Et3B (3 equiv.), air, CH2Cl2 (0.1 M), 0 °C | 76c) | 0 | 0 |
| 4 | 3a | COSePh | n-Bu3SnH (2 equiv.), V-40 (40 mol%) | 55 | 4.4 | 0 |
| toluene (0.02 M), reflux | ||||||
| 5 | 3b | COTePh | Et3B (3 equiv.), air, CH2Cl2 (0.02 M), r.t. | 15 | 49 | 0 |
a) 20–39 mg scale. b) Yield calculated from 1H-NMR using CH2Br2 as an internal standard. c) Isolated yield.
As a base was not required, the radical generating conditions for selenide 2a and telluride 2b were applicable to the coupling with 4b (Table 2). The α-alkoxy radical was formed from selenide 2a with n-Bu3SnH and V-40 and stereospecifically added to 4b to afford the adduct 5b (entry 2). Because 5b was obtained as a diastereomeric and keto-enol tautomeric mixture, the yield was determined after its conversion to acetyl vinyl ether 7 (58% over two steps). Alternatively, telluride 2b and 4b were converted into 5b by the action of Et3B and air in CH2Cl2 at 0 °C (entry 3), and 7 was produced in 76% yield after the acetylation. Therefore, 2a and 2b functioned as the coupling partners of both 4a and 4b, corroborating their high utility.
The reagent systems used in entries 2 and 3 were then applied to acyl selenide 3a and acyl telluride 3b, respectively, in the presence of 4b (Table 2). The acetylated adduct 7 was generated in 55% yield through the reaction of acyl selenide 3a, 4b, n-Bu3SnH, and V-40 in refluxing toluene, followed by acetylation (entry 4). Acetylated adduct 8 with the retained carbonyl group was also obtained as a minor byproduct (4.4%). Interestingly, the ratio of 7 and 8 was reversed when acyl telluride 3b and 4b were subjected to Et3B in CH2Cl2 under air at room temperature (entry 5). The Et3B/O2-promoted coupling and ensuing acetylation gave rise to ketone 8 in 49% yield along with 7 in 15% yield. Therefore, the combination of radical precursor 3b and radical acceptor 4b altered the major pathway from the addition of the α-alkoxy radical to the addition of the acyl radical.
The reaction pathways of radical species generated from 1, 2a/2b, and 3a/3b are summarized in Chart 3. K2HPO4 deprotonates carboxylic acid 1 to afford the corresponding carboxylate, which undergoes one-electron oxidation by photo-activated B to form carboxyl radical 8. The spontaneous CO2 elimination readily converts 8 into α-alkoxy bridgehead radical 9. Selenide 2a and telluride 2b are transformed into the same radical 9 via homolytic cleavage of the C–Se and C–Te bonds, respectively, while the C–Se and C–Te cleavage reactions of acyl selenide 3a and acyl telluride 3b provide α-alkoxyacyl radical 10. CO expulsion from 10 is significantly slower than CO2 expulsion from 8 (k = 1.1 × 1010 s−1 for (CH3)3CCO2· vs. k = 1.4 × 106 s−1 for (CH3)3CCO·),33–36) and the neighboring α-oxygen atom at the bridgehead position does not facilitate C–C=O scission because of the weak interaction between the C–C=O σ* orbital and the oxygen lone pair. Nevertheless, decarbonylation occurs upon heating (110 °C), and 5aa/5ab (entry 7, Table 1) and 5b (entry 4, Table 2) are generated from acyl selenide 3a. When acyl telluride 3b is used at room temperature, however, the decarbonylation is further decelerated, and the yields of the acyl radical adducts 6aa/6ab (entry 8, Table 1) and 6b (entry 5, Table 2) are increased. As the intermolecular reaction between 10 and 4b with the extra electron-withdrawing cyanide is faster than that between 10 and 4a, 6b is mainly produced prior to the CO elimination from 10. Therefore, the present study clarified that the product ratio between 5a/5b and 6a/6b was controlled by the eliminating group (CO2 vs. CO), the reaction temperature (110 °C vs. room temperature), and the acceptor electrophilicity (4a vs. 4b).

In summary, we comprehensively compared intermolecular radical coupling reactions of trioxaadamantyl carboxylic acid 1, selenide 2a, telluride 2b, acyl selenide 3a, and acyl telluride 3b with 4-TBSoxy-cyclopent-2-en-1-one (4a) and 5-oxo-1-cyclopentene-1-carbonitrile (4b). Carboxylic acid 1, selenide 2a, and telluride 2b coupled with 4a under the individually optimized conditions, leading to high-yielding and stereoselective formation of 5aa. Compared with 1, 2a and 2b, 3a and 3b were less effective coupling partners with 4a. Because of the more base-labile and electrophilic nature of 4b, the reaction outcomes using 4b differed from those using 4a. While the basic and photoredox reaction conditions decomposed 4b, compounds 2a, 2b, and 3a functioned as efficient precursors of the α-alkoxy bridgehead radical for coupling with 4b. In contrast, the acyl radical generated from 3b directly added to the olefin of 4b, producing 6b with the retained carbonyl group. Therefore, the obtained results together uncovered the important factors for enabling a high-yielding intermolecular coupling and controlling the decarbonylation. We hope that the present data will provide a guide for selecting radical precursors and acceptors, and for designing retrosyntheses of highly oxygenated natural products.
This research was financially supported by Grants-in-Aid for Scientific Research (S) (JP22H04970) and Transformative Research Areas “Green Catalysis Science” (JP24H01838) to M.I., and Scientific Research (C) (JP23K04749) to K.H. from JSPS. X-ray crystallographic analyses were supported by Advanced Research Infrastructure for Materials and Nanotechnology in Japan of MEXT (JPMXP1224UT0044).
The authors declare no conflict of interest.
This article contains supplementary materials.