Results and Discussion
We designed 1 as a fluorescence probe for detecting NTR activity based on the sr-TICT mechanism (Fig. 2). The probe possesses a methyl group at the 2-position and both p-nitrobenzyl and ethyl groups on the nitrogen atom at the 3-position of the xanthene moiety in a rhodamine scaffold. We expected that intramolecular steric repulsion between the methyl and nitrobenzyl groups on 1 would induce fluorescence quenching through the sr-TICT mechanism. NTR can reduce an aromatic nitro group to the corresponding amino group,11,12) and the nitrobenzyl group of 1 was designed to be spontaneously released as iminoquinone methide after the enzymatic reaction. Thus, upon reduction by NTR, the bulky benzyl group of 1 would be released, generating a highly fluorescent product 2.
Compound 1 and its control compound 3, lacking the nitro group on the benzyl moiety, were synthesized in four steps starting from rhodamine 6G (Chart 1). Briefly, rhodamine 6G was reduced with sodium borohydride (NaBH4) to afford the colorless leuco form of rhodamine 6G (4). Subsequently, the leuco intermediate was reacted with 4-nitrobenzaldehyde or benzaldehyde and borane-2-methylpyridine complex for reductive amination. The products were oxidized to the colored rhodamine form (5 or 6), and the ethyl ester group on the benzene ring was hydrolyzed in a mixture of CH3CN and 2 N NaOH aq., affording 1 or 3.

Chart 1. Synthetic Scheme for 1 and 3
We first examined the photophysical properties of 1–3 (compound 2 is a commercially available rhodamine dye, known as rhodamine 19) (Fig. 3, Table 1). As expected, both 1 and 3 were almost non-fluorescent (Φfl < 0.01), while the expected product from 1 after the enzymatic reaction with NTR, 2, showed strong fluorescence (Φfl = 0.85). We also determined the molar extinction coefficients of 1–3 at the maximum absorption wavelengths in Dulbecco’s phosphate-buffered saline (DPBS) (pH 7.4). The molar extinction coefficients of 1 and 3 were smaller than that of 2, probably because of intramolecular spirocyclization due to the electron-withdrawing property of the p-nitrobenzyl or benzyl group.
Table 1. Photophysical Properties of
1–
3 | λabs (nm) | λem (nm) | ε (M−1·cm−1) | Φfl |
|---|
| 1 | 528 | 548 | 19000 | <0.01 |
| 2 | 522 | 548 | 81000 | 0.85 |
| 3 | 536 | 557 | 61000 | <0.01 |
Since 1 possesses a nitro group, a strongly electron-withdrawing group, near the fluorophore, it was expected that a fluorescence quenching mechanism termed donor-excited photoinduced electron transfer (d-PeT) might occur.13,14) But, based on the negligible fluorescence quantum yield observed not only for 1 but also 3 without a nitro group (Table 1), it seems that the nitro group is not necessary for the fluorescence quenching. To further verify the fluorescence quenching mechanism of 1, its absorption and fluorescence spectra were examined in solutions with increasing glycerol concentrations. Since the formation of the TICT state requires intramolecular rotation, it is generally the case that the fluorescence intensity increases with increasing solution viscosity, i.e., higher glycerol concentration.7,15) When the glycerol concentration was increased from 0 to 90%, the fluorescence quantum yield of 1 increased from 0.01 to 0.21, as expected (Supplementary Fig. S2b), though an absorbance increase also occurred, probably because intramolecular spirocyclization of 1 is easier in solutions with lower glycerol concentrations (Supplementary Figs. S2a, c). These results suggest that the fluorescence quenching of 1 is mainly due to the sr-TICT mechanism.
We then examined whether 1 could detect the enzymatic activity of NTR. For this purpose, we employed a commercially available NTR from Escherichia coli, NfsB, which is a nicotinamide adenine dinucleotide (NADH)-dependent oxygen-insensitive type I NTR.16,17) Upon incubation of 1 with NfsB and NADH in DPBS, the fluorescence intensity of 1 exhibited a time-dependent increase of up to approximately 15-fold, reaching a plateau after 60 min (Fig. 4). On the other hand, when the heat-inactivated enzyme was added to the solution of 1, little fluorescence increase was observed (Supplementary Fig. S3). Thus, 1 successfully detected the enzymatic activity of NTR. The maximum absorbance also increased approximately 4-fold with a blue shift of the maximum absorption wavelength from 528 to 522 nm during the reaction. These results match those in Fig. 3 and Table 1, and support the occurrence of the N-debenzylation from 1 to 2, as shown in Fig. 2. We further analyzed the enzymatic reaction product of 1 by HPLC. After incubation with NfsB and NADH in DPBS (pH 7.4) at 37 °C for 1 h, 1 was completely converted to 2, as shown in Fig. 5. This further supports the occurrence of the expected reaction of 1 with NTR, affording the highly fluorescent product 2. We next examined the kinetics of the enzymatic reaction of 1 with NfsB to produce 2. The apparent kcat/Km value was 920 M−1 s−1, which is relatively slow compared with reported substrates (Supplementary Fig. S4, Supplementary Table S1), probably because the iminoquinone methide is eliminated from the amino group of the xanthene moiety of 1 whereas it is normally released from carboxy or phenol moieties with relatively low pKa values, which represent excellent leaving groups.9,10) Nevertheless, the fluorescence increase of 1 upon the enzymatic reaction was sufficiently large and fast to demonstrate the feasibility of developing sr-TICT-based fluorescence probes utilizing quinone methide chemistry.
In conclusion, we designed and synthesized 1 as a fluorescence probe to detect NTR activity based on the sr-TICT mechanism. This molecular design successfully applies azaquinone methide elimination, which is widely used as a fluorescence turn-on switch for various enzyme activities such as peptidase and reductase, to construct an sr-TICT-based fluorescence probe. In contrast to conventional design strategies that control the fluorescence properties of rhodamine dyes through spirocyclization,3,18) the sr-TICT mechanism offers the advantage that the intrinsic cationic charge of the rhodamine scaffold is retained after the enzymatic reaction, so that the fluorescence probes are expected to be localized in mitochondria in living cells.19) Indeed, we verified that 1 was specifically localized in mitochondria by means of live-cell fluorescence imaging based on probably a weak fluorescence of unreacted 1 (Supplementary Fig. S5). Thus, probes based on this mechanism are expected be useful for detecting enzymatic activity in mitochondria. We believe our mechanism based on simple structural “twisting” should enable the development of new types of fluorescence probes.
Experimental
General Procedure and MaterialsReagents and solvents were of the highest grade available, purchased from Tokyo Chemical Industries (Tokyo, Japan), Wako Pure Chemical Corporation (Osaka, Japan), Aldrich Chemical Co. (Gillingham, U.K.), Dojindo (Kumamoto, Japan), Sigma-Aldrich (St. Louis, MO, U.S.A.), and Invitrogen (Waltham, MA, U.S.A.), and were used without further purification. Reactions were monitored by TLC and electrospray ionization (ESI) mass spectrometry, and all compounds were purified using medium-pressure liquid chromatography (MPLC) and/or preparative HPLC. NTR from E. coli, NfsB, was purchased from Sigma-Aldrich. Rhodamine 19 (2) was purchased from Tokyo Chemical Industries. Human lung carcinoma cell line A549 was purchased from RIKEN Bioresource Center cell bank (Tsukuba, Japan). Dulbecco’s phosphate-buffered saline (DPBS), minimum essential medium (MEM), MEM (no glutamine, no phenol red), fetal bovine serum (FBS), non-essential amino acid (NEAA), and CellLight™ mitochondria-RFP were products of Thermo Fisher Scientific (Waltham, MA, U.S.A.). Poly-lysine-coated glass-bottom dishes were purchased from Matsunami Glass Ind. (Osaka, Japan).
InstrumentsNMR spectra were recorded on a Bruker AVANCE500 instrument at 500 MHz for 1H-NMR and at 125 MHz for 13C-NMR. Mass spectra (ESI+ or ESI−) were measured with a JEOL JMS-T100LP AccuTOF (Tokyo, Japan). MPLC purification was performed using an EPCLC AI-580S flash chromatography instrument (Yamazen, Osaka, Japan) for normal-phase chromatography and/or an Isolera One flash chromatography instrument (Biotage, Uppsala, Sweden) for reversed-phase chromatography. HPLC purifications were performed on an HPLC system composed of a pump (PU-4580, JASCO, Tokyo, Japan) and a detector (MD-4010 or FP-2020, JASCO) equipped with a reversed-phase column, Inertsil ODS-3 (10 × 250 mm) (GL Sciences Inc., Tokyo, Japan), using eluent A (H2O containing 0.1% trifluoroacetic acid (TFA) (v/v)) and eluent B (CH3CN containing 0.1% TFA (v/v)), at the flow rate of 5.0 mL/min. HPLC analyses were performed on a system composed of a pump (PU-4580, JASCO) and a detector (MD-4010, JASCO), equipped with a reversed-phase column, Inertsil ODS-3 (4.6 × 250 mm) (GL Sciences Inc.), using eluent A and eluent B at the flow rate of 1.0 mL/min. Absorption spectra were obtained with a UV-2550 UV-Vis spectrophotometer (Shimadzu, Kyoto, Japan). Fluorescence spectroscopic studies were performed with an F-7000 fluorescence spectrophotometer (Hitachi, Tokyo, Japan). The excitation and emission slit widths were 2.5 nm for both excitation and emission. The photomultiplier voltage was set at 400 V. Absolute fluorescence quantum yields were determined with a Quantaurus QY absolute PL quantum yield spectrometer (Hamamatsu Photonics, Shizuoka, Japan). Fluorescence confocal microscopic images were acquired using an FV3000 confocal laser scanning microscope (Olympus, Tokyo, Japan).
Determination of Molar Extinction Coefficients and Fluorescence Quantum YieldsThe molar extinction coefficients of 1, 2, and 3 were determined according to the following procedure. A solution of dimethyl sulfoxide (DMSO)-d6 containing N,N-dimethylacetamide (DMAc) was divided into four portions, and several milligrams of fluorescein, 1, 2 or 3 was added to each portion. The 1H-NMR spectrum of each solution was measured, and the integrated value of a peak corresponding to a methyl group of DMAc (3H) was compared with that of an appropriate peak of each compound. Based on the ratio of the integrated values, the molar ratio of DMAc to each compound in the solution was determined. Next, the NMR solution containing fluorescein (3.0 µL) was diluted with 0.01 N NaOH aq. (3.0 mL), and the absorption spectrum was measured. The concentration of fluorescein was determined based on the absorbance at 490 nm and the reported molar extinction coefficient in 0.01 N NaOH aq. (ε490 = 88000).20) The concentration of DMAc in the original solution was estimated based on the concentration of fluorescein and the molar ratio of fluorescein to DMAc in the solution. Similarly, the NMR solutions containing 1, 2, and 3 (3.0 µL each) were diluted in DPBS (pH 7.4) (3.0 mL each), and the absorption spectrum of each solution was measured. The concentration of each compound was calculated based on the concentration of DMAc and the molar ratio of each compound to DMAc in the solution. Finally, the molar extinction coefficients of 1, 2, and 3 at their maximum absorbance wavelengths were determined based on the corresponding absorbance and their concentrations in the solution. The fluorescence quantum yields of 1, 2, and 3 in DPBS (pH 7.4) containing 0.1% DMSO as a cosolvent were determined using an absolute quantum yield spectrometer.
Absorption and Fluorescence Spectra in DPBS Containing Various Glycerol ConcentrationsSolutions of DPBS (pH 7.4) containing 0–90% (w/v) glycerol were incubated in a thermostated chamber at 25 °C for 30 min before addition of 1 (final 0.2 µM). Each solution was mixed using a vortex mixer, and air bubbles were removed by centrifugation. The solution was transferred to a quartz cuvette (1 × 1 cm), and the absorption and fluorescence spectra excited at 470 nm were measured.
Enzymatic AssayDPBS (pH 7.4) in a quartz cuvette (1 × 1 cm) was pre-incubated in a thermostated chamber at 37 °C for 30 min. To the buffer, 1 (3 µM) and NfsB (10 µg/mL) were added with 0.1% DMSO as a cosolvent. Then, NADH (final 0.5 mM) was added to initiate the reaction. For the reaction with the heat-inactivated enzyme, the NfsB solution (30 µg in 100 µL in DPBS) was heated at 80 °C for 10 min. After cooling, the entire solution including any precipitate was added to the assay solution. The enzymatic reaction was monitored every 10 min with absorption and fluorescence spectrophotometers. Alternatively, 1 (1 µM) was incubated in DPBS (pH 7.4) with 0.5 mM NADH and NfsB (10 µg/mL) at 37 °C for 60 min. The reaction mixture was passed through a 0.45 mm filter and analyzed using HPLC.
Kinetic AssayA calibration curve of fluorescence intensity versus concentration of 3 (the enzymatic reaction product of 1) was created by measuring the fluorescence intensity of 0.02, 0.1, 0.4, 0.7, and 1.0 µM 3 in 10 mM Tris–HCl (pH 7.0) at 25 °C. The enzyme reaction for the kinetic study was performed in 10 mM Tris–HCl (pH 7.0) containing NADH (0.5 mM), NfsB (10 µg/mL) and 1 (0.02–10 µM) with DMSO (<0.1%) as a cosolvent at 25 °C. The reaction was initiated upon the addition of NADH, and the change in fluorescence intensity was monitored (excitation wavelength: 520 nm, emission wavelength: 548 nm). The initial velocity was calculated from the change in fluorescence intensity with the calibration curve, and plotted against substrate concentration. The results were fitted to the Michaelis–Menten equation (1) using KaleidaGraph software to calculate the apparent kinetic parameters.
 | (1) |
where V = initial velocity and [S] = substrate concentration.
kcat was derived from the following equation (2).
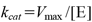 | (2) |
where [E] = molar enzyme concentration (0.42 µM), which was calculated using the molecular weight of NfsB (M.W. = 23904).
Culture Conditions of Cells and Transfection MethodA549 cells were cultured in MEM containing 10% FBS, 0.1 mM NEAA and 1% penicillin–streptomycin at 37 °C under 5% CO2 in air. For transfection, A549 cells (5 × 104 cells/cm2) were seeded on a 35 mm poly-lysine-coated glass-bottom dish, and CellLight™ mitochondria-RFP reagent (25 µL/mL) was added. The cells were cultured for 24 h before the assay.
Fluorescence Confocal MicroscopyA549 cells (5 × 104 cells/cm2) were seeded on a 35 mm poly-lysine-coated glass-bottom dish and cultured for one day before the assay. The culture medium was exchanged for 2 mL MEM (no glutamine, no phenol red) containing 1 (3 µM) with 0.1% DMSO as a cosolvent. The cells were incubated in the medium containing 1 for 4 h, then washed with 1 mL DPBS three times, and 2 mL MEM (no glutamine, no phenol red) was added. Images were acquired using a confocal laser scanning microscope with excitation and emission wavelengths of 488 and 500–540 nm for 1, and 561 and 600–670 nm for RFP, respectively.
Synthesis of 4Rhodamine 6G (684 mg, 1.43 mmol) was dissolved in MeOH (20 mL), and NaBH4 (1.08 g, 28.0 mmol) was slowly added to the solution at 0 °C. The mixture was stirred for 30 min at room temperature, then water was added to quench the reaction. The mixture was evaporated to remove MeOH, and the residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using MPLC (NH silica, hexane/CH2Cl2 = 80/20 to 50/50) to afford 4 (573 mg, 1.29 mmol, y. 90%). 1H-NMR (500 MHz, CD2Cl2) δ: 1.31 (t, 6H, J = 7.1 Hz), 1.46 (t, 3H, J = 7.2 Hz), 1.98 (s, 6H), 3.25 (q, 4H, J = 7.2 Hz), 3.43 (br s, 2H), 4.66 (q, 2H, J = 7.1 Hz), 6.00 (s, 1H), 6.32 (s, 2H), 6.72 (s, 2H), 7.07 (dd, 1H, J = 7.9, 1.0 Hz), 7.16 (dt, 1H, J = 7.9, 0.8 Hz), 7.74 (dd, 1H, J = 7.9, 1.3 Hz); 13C NMR (125 MHz, CD3OD) δ: 14.3, 14.7, 16.7, 37.4, 38.7, 61.4, 97.2, 112.7, 117.3, 125.6, 129.1, 129.9, 130.7, 131.4, 132.0, 146.3, 149.8, 150.4, 168.7; HR-MS (ESI+): Calcd for [M + H]+, 445.2491. Found, 445.2481 (−1.0 mmu).
Synthesis of 1Compound 4 (451 mg, 1.01 mmol) and 4-nitrobenzaldehyde (186 mg, 1.21 mmol) were dissolved in THF (8 mL) and the solution was stirred for 10 min at room temperature, then conc. H2SO4 (1 drop) was added, and stirring was continued for an additional 5 min. Borane-2-methylpyridine complex (522 mg, 4.09 mmol) was added to the mixture and stirring was continued for 24 h at room temperature. Chloranil (2.20 g, 8.76 mmol) was added, and the mixture was further stirred for 10 min at room temperature. Then, 2 N NaOH aq. was added to basify the solution, and the organic phase was collected and evaporated. The residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using reversed-phase MPLC sequentially under the following two conditions: (i) A/B = 80/20 to 0/100 linear gradient (solvent A: H2O containing 100 mM triethylammonium acetate; solvent B: CH3CN containing 100 mM triethylammonium acetate), and (ii) A/B = 80/20 to 0/100 linear gradient (solvent A: H2O containing 0.1% TFA; solvent B: CH3CN containing 0.1% TFA), to afford crude 5 (482 mg). The crude 5 (280 mg) was dissolved in a mixture of CH3CN (10 mL) and 2 N NaOH aq. (20 mL). The solution was stirred for 150 min at 50 °C, then cooled to room temperature, and acidified by adding 2 N HCl aq. The residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using MPLC (silica gel, MeOH/CH2Cl2 = 20/80) to afford crude 1 (62.6 mg). This was dissolved in a mixture of CH3CN (10 mL) and 2 N NaOH aq. (20 mL). The solution was stirred for 80 min at 50 °C, then cooled to room temperature, and acidified by adding 2 N HCl aq. The residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using MPLC (silica gel, MeOH/CH2Cl2 = 20/80) and further purified by reversed-phase HPLC under the following conditions: A/B = 80/20 to 0/100 linear gradient (solvent A: H2O containing 0.1% TFA; solvent B: CH3CN containing 0.1% TFA). The obtained fraction was neutralized with NH3 aq., and the solution was evaporated to remove CH3CN and lyophilized to afford 1 (61.2 mg, 111 µmol, y. 19% in 3 steps). 1H-NMR (500 MHz, CD3OD with one drop of CD3CO2D) δ: 1.30 (t, 3H, J = 7.1 Hz), 1.41 (t, 3H, J = 7.2 Hz), 2.17 (s, 3H), 2.37 (s, 3H), 3.46 (q, 2H, J = 7.1 Hz), 3.60 (q, 2H, J = 7.2 Hz), 4.76 (s, 2H), 6.98 (s, 1H), 7.00 (s, 1H), 7.04 (s, 1H), 7.31 (s, 1H), 7.38 (dd, 1H, J = 6.2, 1.5 Hz), 7.60 (d, 2H, J = 8.3 Hz), 7.82 (td, 1H, J = 6.2, 1.5 Hz), 7.86 (td, 1H, J = 6.2, 1.5 Hz), 8.20 (d, 2H, J = 8.3 Hz), 8.37 (dd, 1H, J = 6.2, 1.1 Hz); 13C NMR (125 MHz, CD3OD with one drop of CD3CO2D) δ: 12.8, 13.9, 17.5, 20.5, 39.8, 55.7, 94.9, 107.4, 117.4, 117.5, 124.8, 129.3, 129.7, 130.6, 131.4, 131.6, 132.1, 132.2, 132.3, 132.6, 134.0, 135.2, 146.9, 148.7, 155.6, 159.9, 160.2, 160.4, 161.3, 168.1, 173.5; HR-MS (ESI+): Calcd for [M + H]+, 550.2342. Found, 550.2352 (+1.0 mmu).
Synthesis of 3Compound 4 (270 mg, 0.607 mmol) and benzaldehyde (0.725 mL, 7.03 mmol) were dissolved in THF (20 mL) and the mixture was stirred for 10 min at room temperature. conc. H2SO4 (3 drops) was added to the mixture, and stirring was continued for an additional 5 min. Borane-2-methylpyridine complex (504 mg, 4.01 mmol) was added and stirring was continued for 24 h at room temperature. Chloranil (1.76 g, 7.03 mmol) was added, and the mixture was stirred for 10 min at room temperature and acidified by adding 2 N NaOH aq. The organic phase was evaporated and the residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using reversed-phase MPLC under the following conditions: A/B = 99/1 to 0/100 linear gradient (solvent A: H2O containing 0.1% TFA; solvent B: CH3CN containing 0.1% TFA), to afford crude 6 (364 mg). The crude 6 (328 mg) was dissolved in a mixture of CH3CN (10 mL) and 2 N NaOH aq. (20 mL), and the solution was stirred for 150 min at 50 °C, then cooled to room temperature, and acidified by adding 2 N HCl aq. The residue was extracted with CH2Cl2. The organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using MPLC (silica gel, MeOH/CH2Cl2 = 20/80) to afford crude 3 (35.2 mg). This was dissolved in a mixture of CH3CN (10 mL) and 2 N NaOH aq. (20 mL) and the solution was stirred for 80 min at 50 °C, then cooled to room temperature, and acidified by adding 2 N HCl aq. The solution was extracted with CH2Cl2, and the organic phase was dried over Na2SO4 and evaporated to dryness. The residue was purified using MPLC (silica gel, MeOH/CH2Cl2 = 20/80) and further purified by reversed-phase HPLC under the following conditions: A/B = 80/20 to 0/100 linear gradient (solvent A: H2O containing 0.1% TFA; solvent B: CH3CN containing 0.1% TFA). The obtained fraction was neutralized with NH3 aq., and the solution was evaporated to remove CH3CN and lyophilized to afford 3 (21.2 mg, 42 µmol, y. 8% in 3 steps). 1H-NMR (500 MHz, CD3OD with one drop of CD3CO2D) δ: 1.29 (t, 3H, J = 7.1 Hz), 1.37 (t, 3H, J = 7.2 Hz), 2.16 (s, 3H), 2.36 (s, 3H), 3.46 (q, 2H, J = 7.1 Hz), 3.59 (q, 2H, J = 7.2 Hz), 4.66 (s, 2H), 6.97 (s, 1H), 7.00 (s, 1H), 7.02 (s, 1H), 7.24 (m, 1H), 7.28 (s, 1H), 7.32 (br s, 2H), 7.33 (br s, 2H), 7.39 (dd, 1H, J = 6.2, 1.4 Hz), 7.82 (td, 1H, J = 6.2, 1.4 Hz), 7.86 (td, 1H, J = 6.2, 1.4 Hz), 8.34 (dd, 1H, J = 6.2, 1.4 Hz); 13C NMR (125 MHz, CD3OD with one drop of CD3CO2D) δ: 12.9, 13.9, 17.5, 20.8, 39.8, 56.4, 94.9, 106.6, 117.0, 117.1, 128.5, 128.7, 128.8, 129.8, 130.5, 131.4, 131.6, 131.8, 132.1, 132.4, 132.6, 134.0, 135.3, 138.7, 156.0, 159.5, 160.2, 160.9, 161.2, 168.1, 173.5; HR-MS (ESI+): Calcd for [M + H]+, 505.2491. Found, 505.2527 (+3.6 mmu).




