Review
Unconventional Synthetic Approaches to Unusual Peptide Derivatives
2025 Volume 73 Issue 4 Pages 268-282
Details
2025 Volume 73 Issue 4 Pages 268-282
Peptides that contain unusual motifs, such as non-proteinogenic amino acids (AAs) and/or macrocyclic substructures, have recently attracted great attention as a new modality in medium-sized-molecule drug discovery. Therefore, it is highly important to develop methods for the chemical synthesis of a wide variety of such unusual peptide derivatives, which are often difficult to prepare via conventional synthetic approaches. In this review, the development of unconventional approaches for the synthesis of unusual peptide derivatives is discussed. Specifically, a novel external-oxidant-mediated decarboxylative condensation of α-ketoacids that can be applied to the synthesis of a wide variety of unusual peptide derivatives is reported. Moreover, an organocatalytic asymmetric Mannich-type addition is discussed that provides chiral β-amino-α-ketoacids, which are required as starting materials for the decarboxylative condensation. In this reaction, the adducts corresponding to various unusual AA side chains are obtained in high yield and excellent stereoselectivity. Furthermore, the “N-chloropeptide strategy” is proposed as a new method for the chemical modification of peptides without the need for a reactive AA residue.
Peptides are biopolymers derived from amino acids (AAs) that play a wide variety of roles in the human body. Recently, unusual peptides, that is, peptides that contain, for example, non-proteinogenic AAs and/or macrocyclic substructures, with molecular weights of up to approximately 2000, have been recognized as an important modality in medium-sized-molecule drug discovery.1) Conventionally, it has been difficult to apply peptides as pharmaceuticals due to their low stability in vivo and poor membrane permeability; however, macrocyclic peptides such as naturally occurring cyclosporin A, with high stability and binding affinity to their targets, have been discovered as promising drug candidates. In this context, the introduction of unusual motifs such as non-proteinogenic AAs and/or macrocyclic skeletons into peptides is an important strategy for improving their pharmacological activity and metabolic stability. Thus, the development of efficient methods for the synthesis of unusual peptide derivatives is an urgent issue in synthetic organic chemistry. 9-Fluorenylmethyloxycarbonyl (Fmoc)-based solid-phase peptide synthesis (SPPS) has been established as a reliable method for the synthesis of small amounts of specific common peptide sequences, mostly consisting of proteinogenic AAs; if the corresponding AAs are available, these sequences can easily be obtained via a linear synthetic route involving sequential elongation with individual AAs in the N-terminal direction. On the other hand, some unusual structures require multiple steps for the preparation of AAs, and some structures are not suitable for the sequential-elongation approach.
Against this background, our group has developed new synthetic methods for the synthesis of unusual peptide derivatives that contribute to peptide drug discovery (Chart 1). Specifically, we have developed a novel external-oxidant-mediated decarboxylative condensation of α-ketoacids that can be applied to the synthesis of a wide variety of unusual peptide derivatives (Chart 1a). In addition, we have developed an organocatalytic asymmetric Mannich-type addition to provide the chiral β-amino-α-ketoacids that are required as starting materials in the decarboxylative condensation, and succeeded in obtaining the adducts corresponding to various unnatural AA side chains in high yield and excellent stereoselectivity (Chart 1b). Furthermore, we have proposed the “N-chloropeptide strategy” as a new method for the chemical modification of peptides that does not require reactive AA residues (Chart 1c). In this review, the series of investigations that led to the development of these unconventional approaches for the synthesis of unusual peptide derivatives are presented.

The most common method for the formation of amide bonds is dehydrative condensation, and various condensation agents for the synthesis of peptides have been developed.2) However, attempts to couple peptide fragments rather than individual AAs for more convergent synthetic approaches have often failed due to epimerization caused by the formation of undesirable azlactones. Despite the development of various epimerization inhibitors, fragment condensation remains problematic. Our group has argued that there may be no fundamental solution to this problem as long as the formation of the peptide bond proceeds through active-ester intermediates. Thus, we decided to momentarily set aside practicality concerns and search for an alternative to the well-established dehydrative-condensation approach to form peptide bonds, focusing on α-ketoacids as a potentially useful acylating agent.
α-Ketoacids are interesting motifs that can be considered aldehyde equivalents by decarboxylation. In biochemistry, α-ketoacids such as pyruvic acid can be converted to Breslow intermediates with the assistance of thiamine pyrophosphate; subsequent oxidative transformation provides acyl-CoAs, which are important metabolic acylating agents.3) In contrast, organic reactions using α-ketoacids have traditionally remained underexplored. In recent years, acylation accompanied by decarboxylation has been intensively investigated. In 2006, Bode et al. made a pivotal breakthrough by developing ketoacid-hydroxylamine ligation using hydroxylamines.4) This amidation protocol involves the simple mixing of 2 fragments without any reagents, produces only CO2 and H2O as byproducts, and it has already been applied to the chemical ligation of peptides, taking advantage of its bioorthogonality. However, given that the preparation of the nucleophile hydroxylamine is necessary, this method is not suitable for routine peptide condensation. Against this background, we envisioned the development of new decarboxylative transformations of α-ketoacids using an external oxidant with high functional-group tolerance, which are outlined below (Chart 2).
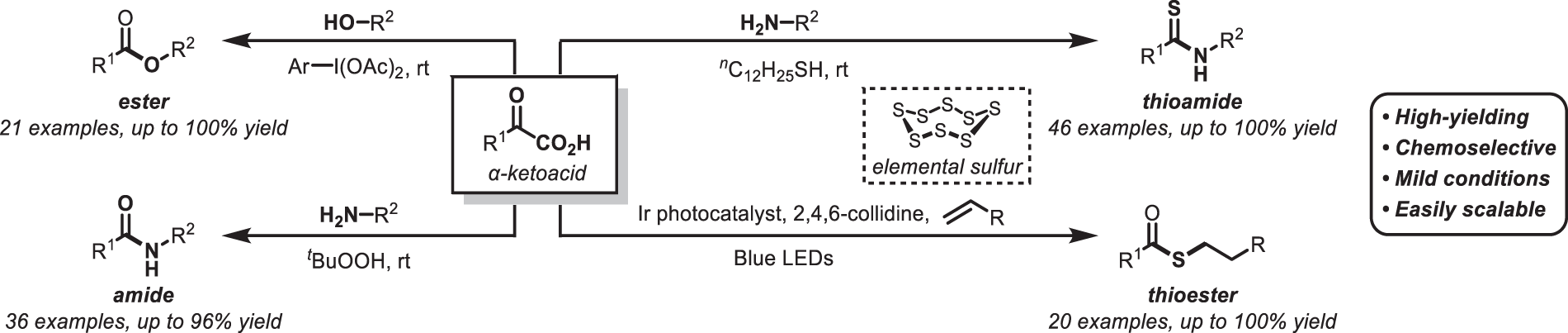
First, we reasoned that α-ketoacids could potentially be converted into reactive acylating agents using hypervalent iodine (III) reagents, and that this could potentially enable a wide range of subsequent oxidative transformations. Based on this concept, we screened various nucleophiles and reaction conditions and succeeded in the decarboxylative acylation of alcohols as relatively unreactive nucleophiles.5) Treatment of an alcohol and α-ketoacid with 1-(diacetoxy)iodo-3-nitrobenzene (1) at room temperature in CH2Cl2 provided the corresponding esters in excellent yield (Table 1). Primary and secondary alcohols can be applied in this reaction, and both acyclic and cyclic alcohols provided the corresponding esters (2–4) in excellent yield. It should be noted here that the reaction rates are influenced significantly by the steric environment of the alcohols, whereby tertiary alcohols fail to furnish the desired esters. Despite the uniqueness of these oxidative-acylation conditions, electron-rich benzene and benzyl alcohol were fully tolerated in this reaction, and the corresponding esters (5, 6) were obtained in 95 and 96% yield, respectively, without any undesirable oxidation. While hypervalent iodine (III) species can potentially react with unsaturated bonds, they did not damage the terminal olefin and alkyne functionality of 7 and 8, for which the esterification successfully proceeded. We then applied the esterification to alcohols that bear electrophilic sites, which revealed that allyl ester, thioester, alkyl halide, and aldehyde substrates were tolerated under the mildly acidic reaction conditions, that is, esters 9–12 were obtained without significant degradation. Heterocycles were also applicable, that is, oxindole- and pyridine-containing esters 13 and 14 were obtained in 90 and 82% yield, respectively. Then, we examined the scope of the α-ketoacids (15–18). Substitution adjacent to the carbonyl group did not have a significant influence on the yield of ester 15. The reaction of benzoylformic acid afforded benzoyl ester 16 in 83% yield. We also employed para-substituted substrates to test the electronic effect of the carbonyl group; electron-withdrawing substituents were found to slightly improve the yield of esters 17 and 18.
 |
 |
 |
Alcohol (0.2 mmol), α-ketoacid (0.3 mmol), and 1 (0.3 mmol) were employed in CH2Cl2 (1 mL) at room temperature. Isolated yields are shown.
We also attempted to determine the unique features of this decarboxylative esterification (Chart 3). The ability to achieve selective chemical modifications through the introduction of AA or peptide structures would represent a powerful tool in medicinal chemistry, and the preservation of stereochemical information is critically important in terms of achieving a practical coupling method. The decarboxylative esterification of Fmoc-Leu-CO2H (19) proceeded successfully to afford ester 21 in 91% yield with perfect stereoretention (>99% ee) at a position adjacent to the carbonyl group (Chart 3a). Moreover, we found that the decarboxylative-esterification method is able to differentiate between carboxy groups in the molecules; the reaction conditions predominantly accelerated the conversion of α-ketoacids in the presence of unprotected “normal” carboxy groups. Specifically, we demonstrated the chemoselective esterification of α-ketoglutaric acid 22, which bears both α-ketocarboxy and simple carboxy groups, to afford ester 23 in 61% yield, under preservation of the terminal carboxy group (Chart 3b). This feature also occurred during the selective esterification of hydroxyacid 25, which furnished the corresponding ester (26) in 84% yield, without affecting the unprotected carboxy group.

In order to apply the decarboxylative condensation promoted by an external oxidant to the synthesis of peptides, the formation of an amide bond using amines as a nucleophile was investigated. However, when the above conditions using hypervalent iodine (III) as an oxidant were applied, the oxidation of the amine itself became problematic. In addition, the use of strong electrophiles seriously limits the scope of this method, as peptides contain many side-chain functional groups that are susceptible to oxidation (e.g., thiol, sulfide, amine, and electron-rich aromatic rings). Therefore, we screened for oxidants that could potentially overcome these issues and found that the nucleophilic oxidant t-butylhydroperoxide (TBHP) efficiently promotes the decarboxylative amidation6) (Chart 4). The amidation reaction requires simply an approximately equimolar mixture of the amine and α-ketoacid; it proceeds efficiently at room temperature under neutral conditions and in polar solvents of varying polarity, including N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and EtOH. Thus, the reaction medium can be tailored to the solubility requirements of the substrates.

The optimal amidation protocols were applied to a variety of primary and secondary aliphatic amines to provide the corresponding amides (27–32) in good yield (Table 2). The reaction was also applicable to anilines, and the reaction of p-toluidine afforded anilide 33 in 70% yield, albeit that a longer reaction time was required. Its α-ketoacid scope was also broad, and even sterically hindered α-branched amide 34 and pivalamide 35 were obtained in moderate yield. The benzoylation and formylation products 36 and 37 were also obtained using commercially available α-ketoacids. It should also be noted here that these reaction conditions could be easily carried out on a 10-mmol scale, that is, the use of inexpensive TBHP and α-ketoacid 24 smoothly converted amine 38 into amide 27 in excellent yield without any precautions. The compatibility of various functional groups was investigated, considering the uniqueness of the oxidative acylation conditions. Alkene, alkyne, thioether, and tertiary amine groups did not affect the yield of the amide (39–42). Heterocycles such as furan, thiophene, indole, and pyridine rings were also applicable (43–46). Amidation in the presence of other nucleophilic sites that could potentially be acylated was successful, with the aliphatic amino group reacting preferentially in the presence of unprotected aniline, phenol, and alcohol groups (47–49). Moreover, we discovered that this decarboxylative-amidation protocol can differentiate between different carboxy groups in the same molecule. The decarboxylative esterification and α-ketoglutaric acid and gabapentin chemoselectively afforded the amide in good yield without affecting the unprotected carboxy group (50, 51). Following this initial analysis of the transformation, we attempted the chemoselective acylation of more complex amines. The introduction of hydrophobic motifs, such as fatty-acid chains, in amines can induce dramatic changes in both their biological activity and pharmacokinetics. A fatty-acid chain was easily introduced in good yield onto Baclofen (52), a γ-aminobutyric acid receptor agonist that contains an unprotected carboxy group. Pyridoxamine, which contains phenolic or aliphatic hydroxyl groups and a nucleophilic pyridine moiety, was predominantly acylated on the primary amine nitrogen to provide the corresponding palmitoyl amide (53) in 67% yield.
 |
Amine (1.0 equiv.), α-ketoacid (1.1 equiv.), and TBHP (1.2–1.5 equiv.) in MeCN or DMF at room temperature for 1 h. Isolated yields are shown. a) The reaction proceeded for 12 h. b) α-Ketoacid (1.5 equiv.) and TBHP (2.0 equiv.) were used. c) MeOH was used as the solvent.
We then attempted to apply the novel amidation reaction to the formation of peptide bonds. Preliminary mechanistic studies indicated that the reaction proceeds through an imine intermediate produced by the dehydration of the α-ketoacid and amine; importantly, this means that the amidation does not proceed via the formation of an active ester, which is often converted to an azlactone, which in turn results in epimerization at the α-position (vide supra). We envisioned that this feature of our decarboxylative amidation could make it potentially suitable for the fragment condensation of peptide chains.
Initially, we found that coupling using peptide α-ketoacids that bear a leucine (Leu) residue on the C-terminus proceeded successfully, providing the corresponding peptides in good yield and with good diastereospecificity7) (Chart 4a). Additionally, an N-methyl AA residue was successfully coupled without any epimerization using this fragment-condensation method (Chart 4b, 54); these residues are usually challenging to introduce due to their low reactivity. Moreover, a transformation including α,α-disubstituted AA residues also provided tetrapeptide 55 in excellent yield.8) Despite these remarkable results, we did observe epimerization in 56 when a less-bulky peptide α-ketoacid was used for the decarboxylative amidation (Chart 4c). We attributed this result to imine–enamine tautomerization; however, the precise details were not well understood at that point, and the general applicability of this peptide-bond-forming reaction was also unclear.
First, we investigated the reaction pathway of the decarboxylative amidation. Experimental and theoretical analyses suggested that the reaction proceeded via the formation of an α-imino acid, TBHP addition, and a concerted decarboxylation. We then focused on the relationship between the steric demand of the AA side chains and the ease with which epimerization occurs. Based on the preliminary results shown in Chart 4, we expected the less bulky substituents to be somewhat problematic; however, the reason for this steric effect remained unclear. Therefore, we prepared 3 α-ketoacids derived from alanine (Ala), norvaline (Nva), and Leu (19, 57–58) and 4 AA esters derived from Ala, Nva, valine (Val), and proline (Pro) (59–62) (Chart 5). To systematically examine the steric effects of both the α-ketoacid and amine, we conducted amidation reactions for all possible combinations. The results of the 12 reactions are summarized in Chart 5a and suggest 3 significant trends in the diastereospecificity of the transformation. Firstly, when a less bulky α-ketoacid was used, increased epimerization was observed. Secondly, when a bulkier amine was used, increased epimerization was observed. Thirdly, when Pro was used, no epimerization occurred, regardless of the steric demand of the α-ketoacid.

Based on these systematic examinations of the epimerization, a plausible mechanism for the epimerization was developed (Chart 5b). The degree of epimerization is most likely determined by the relative rates of the TBHP-addition/decarboxylation pathway and the imine–enamine tautomerization. A sterically demanding α-ketoacid side chain would reduce the rate of epimerization by destabilizing the enamine tautomer, while on the other hand, a sterically demanding amine side chain would cause a relative increase in the rate of epimerization by suppressing the competing TBHP-addition/decarboxylation pathway. When a secondary amine such as Pro is used, the addition of TBHP to the cationic iminium intermediate would be extremely fast, while the enamine tautomer would be relatively unstable due to strong intramolecular steric repulsion.
These detailed mechanistic considerations encouraged us to explore modified reaction conditions to enable an increase in the relative rate of the desired TBHP-addition/decarboxylation path. After re-optimizing the reaction conditions, we found that the addition of chloride anions and a slow addition of the α-ketoacid significantly inhibited the epimerization, although the detailed reasons for this improvement are still unclear (Chart 5c). The modified protocol successfully provided the coupling product in good yield and diastereospecificity even in cases when serious epimerization occurred using the original protocol (56, 75).
2.3. Decarboxylative Thioamidation by Mild Activation of Elemental SulfurThioamide is a sulfur-containing functional group whose structure is isoelectronic to that of amides; it can thus be introduced into compounds in the place of amides to improve hydrolysis resistance in vivo. Accordingly, thioamides have been actively investigated as bioisosteres of amides, and synthetic routes to thioamides have also become an important research topic. However, the O–S exchange reaction using Lawesson's reagent has significant problems in terms of reactivity and regioselectivity, and the reaction using thioacylating agents9) also requires multiple reagent-preparation steps.
Against this background, we developed a decarboxylative thioamidation reaction using α-ketoacids.10) A mechanistic analysis of the decarboxylative amidation suggested that the reaction proceeds via the addition of hydroperoxide to an α-iminoacid intermediate. We envisaged that thioamides could potentially be obtained using a nucleophilic sulfurizing oxidant of the type R–Sn–S–H instead of TBHP (Chart 6a). Unfortunately, compared to electrophilic sulfurizing reagents, such nucleophilic reagents are scarce. Accordingly, we planned to activate elemental sulfur using a soft nucleophile such as thiol to generate a nucleophilic sulfurizing species with a Nu–Sn–S–H moiety.

Based on this idea, we investigated the reaction conditions and found that α-ketoacids, amines, and elemental sulfur smoothly couple at room temperature in the presence of alkyl thiol to give the desired thioamide (76) in excellent yield (Chart 6b). The optimal protocol can be applied to the synthesis of a wide variety of thioamide derivatives, and even peptides containing thioamide (77–82) can be prepared by this transformation. Based on the mechanistic analysis of the transformation, we think that the reaction proceeds via the addition of a hydropolysulfide anion generated by the nucleophilic activation of elemental sulfur followed by deprotonation. As this reaction proceeds well with even a catalytic amount of thiol, we concluded that the polysulfide anion is regenerated during decarboxylation. We also found that this reaction is inhibited by a large excess of thiol, probably because the excess thiol promotes the formation of dialkyl polysulfides.
2.4. Visible-Light-Mediated, Decarboxylative Thioesterification Using Elemental SulfurWhile exploring the mild activation of elemental sulfur, we developed thioesterification reactions using elemental sulfur.11) Thioesters are useful carbonyl derivatives that are also used in native chemical ligation,12) and synthetic methods to obtain them have been actively investigated for many years. Current thioester syntheses can be categorized into those using thiols or those using thioacids, albeit that both types require the use of sulfur compounds, which is not desirable in terms of stability and odor.
Against this background, we discovered the first thioesterification method using elemental sulfur as a sulfur source. Considering that the target thioester itself is a reactive functional group, the key to success was the development of a method for the mild activation of stable elemental sulfur. In this context, we focused on the report of Savateev and colleagues from 2018,13) which indicated that elemental sulfur is relatively susceptible to single-electron reduction. We thus envisioned that a photoredox cycle incorporating single-electron oxidation of α-ketoacids could potentially provide a simple approach to constructing thioesters. As a result of the reaction optimization, we found that an olefin, an α-ketoacid, and elemental sulfur can be irradiated with blue light-emitting diode (LED) light in the presence of an iridium photocatalyst and 2,4,6-collidine to afford the desired thioester in excellent yield (Table 3). A wide variety of α-ketoacids are suitable for this transformation, and substrates containing various functional groups such as hydroxy, sulfide, aldehyde, and carboxy groups provided the desired thioesters in good yield (83–88). Bisthioesterification also proceeded very well (89), and the method was expanded to multifunctional complex molecules such as terpenes, steroids, peptides, and unprotected sugars (90–93).
 |
 |
Isolated yields are shown. a) α-Ketoacid (3.0 equiv.), S8 (6.0 equiv.), and 2,4,6-collidine (3.0 equiv.). b) α-Ketoacid (2.5 equiv.), S8 (5.0 equiv.), and 2,4,6-collidine (2.5 equiv.). c) THF was used as the solvent. d) CH2Cl2 was used as the solvent. e) Thioester 93 was obtained in an α : β ratio of 5.3:1, as 3-(D-glucopyranosyl-1-propene (α : β = 5.3:1) was used as the olefinic substrate.
A mechanistic analysis showed that the reaction is triggered by the single-electron oxidation of α-ketoacids and single-electron reduction of elemental sulfur, and that the sulfur species act as a terminal oxidant, a sulfur source, and a hydrogen-atom transfer (HAT) mediator. The multiple roles played by sulfur seem to be crucial to the success and utility of this transformation.
For the decarboxylative transformations described above to be applied to peptide synthesis, the preparation of β-amino-α-ketoacids is essential. In addition, β-amino-α-ketoacids themselves represent an important class of non-proteinogenic AAs that are found in pharmaceuticals and bioactive natural products in the form of the corresponding amides; these molecules covalently bind to nucleophilic moieties such as hydroxy and thiol groups and serve as protease inhibitors due to the highly electrophilic carbonyl group at the α-position (Chart 7a).

Given the utility shown above, several methods for the synthesis of enantio-enriched β-amino-α-ketoacids have been explored to date. In 1994, Wasserman and Ho reported the synthesis of β-amino-α-ketoacids using the 2-step homologation of α-AAs via O3 oxidation of α-acyl cyanophosphorane14) (Chart 7b). The group of Bode has developed a modified protocol using cyanosulfur ylides, which are more easily oxidized by Oxone than phosphorous ylides.15) Despite these groundbreaking studies, practical methods for the synthesis of β-amino-α-ketoacid motifs are still limited to oxidative homologations based on the chiral-pool strategy, which requires readily available α-AAs due to the difficulties associated with constructing small yet highly functionalized chiral motifs. In addition, the derivatization of β-amino-α-ketoacids, including elongation of the peptide chain, is also problematic; an appropriate masking of the carbonyl group, as for example, nitrones or acetals, is essential to avoid undesired degradation and maintain the stereochemical information at the β-position.16) Although the late-stage oxidation of β-amino-α-hydroxy acids and amides is an alternative synthetic approach considering the lability of the α-ketocarboxy derivatives,17) it requires more synthetic steps and has low compatibility with other hydroxy or oxidant-labile functional groups (Chart 7c). Therefore, the development of a method that would enable the rapid preparation of a wide variety of β-amino-α-ketoacids and peptide-elongated derivatives would be highly attractive.
To establish a novel approach allowing the divergent asymmetric synthesis of β-amino-α-ketoacids and their derivatives, we focused on Mannich-type additions, which are used ubiquitously for the construction of AA motifs. Specifically, we envisioned that the stereoselective addition of glyoxylate cyanohydrin, which acts as a masked C2 nucleophile, to N-Boc imines could potentially affords enantio-enriched β-amino-α-ketoacid equivalents and assumed that an amine-based bifunctional organocatalyst could effectively control both the enantio- and diastereoselectivity, as the cyanohydrin possesses several hydrogen-bonding interaction sites. We also expected that the Mannich adducts would be easily converted to β-amino-α-ketoacids that bear various residues including non-proteinogenic ones by sequential deprotections. Moreover, as the cyanohydrin moiety of the Mannich adducts can be regarded as a protecting group for the highly reactive α-carbonyl group, divergent derivatization of the adducts to give peptide α-ketoacids and peptide α-ketoamides could be expected to proceed smoothly7) (Chart 8).

Based on this concept, we started the optimization of the reaction conditions using the asymmetric Mannich-type addition of glyoxylate cyanohydrin to N-Boc imine 95 (Table 4). Nucleophile 94 and N-Boc aliphatic imine 95 were treated with aminobenzothiadiazine catalyst C1, which bears a cyclopentyl substituent and was effective in the preliminary study using N-Boc aromatic imines,18) in toluene at −40°C. The reaction proceeded smoothly, although the adduct was obtained as a mixture of diastereomers in a 7.3:1 ratio and the enantioselectivity of both 96 and 96′ was moderate (entry 1). Interestingly, catalysts C2 and C3, which bear substituents at the 5-position of the aromatic ring, were able to invert the diastereoselectivity while maintaining excellent enantioselectivity; however, the stereoselectivity was still moderate (entries 2 and 3). These unsatisfactory results prompted us to screen other hydrogen-bond donors in the catalyst structure, and we discovered that thiourea catalyst C4 dramatically enhanced both the enantio- and diastereoselectivity to afford adduct 96 in 96% yield as almost a single stereoisomer with 98% ee and 49:1 diastereomeric ratio (entry 4). We also investigated several thiourea catalysts to gain insight into the role of the catalyst structure and found that replacement of the cyclopentyl substituent on the amine moiety with a methyl group lowered the diastereomeric ratio to 7.3:1 while retaining the high enantioselectivity (entry 5). Interestingly, catalysts C6 and C7, which bear different substituents on the aromatic rings of the catalyst, also provided high enantioselectivity and moderate diastereoselectivity (entries 6 and 7). These results indicate that both the steric hindrance of the amine moiety and the electronically negative trifluoromethyl groups at the 3- and 5-positions of the aromatic ring play a crucial role in achieving excellent diastereoselectivity, while the enantioselectivity is controlled only by the structure of the catalyst core.
 |
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (%) | ee (%) | dr(96:96′) | |
| 96 | 96′ | ||||
| 1a) | C1 | 97 | 69 | 71 | 7.3:1 |
| 2a) | C2 | 100 | 69 | 77 | 1:3.3 |
| 3a) | C3 | 95 | 28 | 80 | 1:2.8 |
| 4 | C4 | 96 | 98 | 33 | 49:1 |
| 5 | C5 | Quant. | 95 | 20 | 7.3:1 |
| 6 | C6 | 98 | 93 | 74 | 9.7:1 |
| 7 | C7 | 96 | 93 | 76 | 6.1:1 |
 |
|||||
94 (0.10 mmol), 95 (0.15 mmol), and the catalyst (0.0050 mmol) were stirred in toluene (1.0 mL) at −40°C for 24 h. Isolated yields are shown. Determined by chiral HPLC analysis. a) ent-96 and ent-96′ were obtained as the major enantiomers.
The applicability of the stereoselective Mannich-type addition of glyoxylate cyanohydrin to various N-Boc imines is remarkably broad. (Table 5). It should also be noted here that the reaction can be applied to the direct use of α-amido sulfones as imine precursors if the N-Boc imine is unstable, and 2 protocols can be chosen (Conditions A and B) as the optimal reaction conditions for the Mannich-type addition depending on the stability of the N-Boc imines. Initially, we found that various protecting groups on glyoxylate cyanohydrin were applicable in this reaction without significant changes in the stereoselectivity (96–99). In addition, the synthesis of α-ketoacid equivalents that bear a variety of proteinogenic side chains were applicable (Table 5a), and sterically different aliphatic substituents afforded adducts 100–102 with excellent stereoselectivity. These results prove that the activity of the optimal thiourea catalyst C4 is not significantly influenced by the steric demand of the nucleophile and alkylimine. It should furthermore be noted here that the functionalities in the side chain do not lead to any serious problems in this transformation (103–109). Subsequently, we explored the scope of adducts that bear non-proteinogenic AA side chains (110–121) and found that the reaction proceeded in a highly stereoselective manner in all cases, independent of the size of the substituent and the functional group (Table 5b).
 |
 |
 |
Conditions A: glyoxylate cyanohydrin (1.0 equiv.), N-Boc imine (1.5 equiv.), and thiourea C4 (5 mol%) were stirred in toluene (0.10 M) at –40°C for 24h. Conditions B: glyoxylate cyanohydrin (1.0 equiv.), α-amido sulfone (1.5 equiv.), Cs2CO3 (4.5 equiv.), and thiourea C4 (5 mol%) were stirred in toluene (0.10 M) and H2O (0.10 M) at 0°C for 48h. Isolated yields are shown. Ratios of stereoisomers were determined by chiral HPLC analysis. Conditions are specified in parentheses as (A) or (B). a) Reaction time: 24h.
We investigated in detail the factors that contribute to the appearance of stereoselectivity in the catalytic systems and the excellent results for a wide range of substrates. In addition to the experimental structure–activity relationships of the catalysts and substrates presented above, density functional theory calculations were carefully conducted to investigate the structure of the transition state, and the obtained results suggested that the introduction of a bulky cyclopentyl group on the amine moiety of the catalyst plays an important role in achieving high stereoselectivity. The diastereoselectivity of the addition step is increased by the introduction of a sterically demanding substituent; in addition, 3,5-bis(trifluoromethyl)phenyl groups are also crucial to enhance the diastereoselectivity via the formation of additional noncovalent interactions with the ester moiety of the nucleophile (Chart 9).

We further established the methods for the derivatization of the thus-obtained Mannich-type adducts (Chart 10). In addition to their conversion to β-amino-α-ketoacids via the silver-mediated decomplexation of the cyanohydrin moiety (122–124), we also found a reliable way to prepare peptide α-ketoacids 125, which enabled a decarboxylative peptide coupling with excellent chemoselectivity. Their potential utility as precursors for peptide α-ketoamides was demonstrated via the efficient synthesis of Telaprevir (127) with high optical purity. Importantly, this novel approach represents a highly practical method for the efficient synthesis of bioactive oligopeptides and natural products that contain β-amino-α-ketoacid motifs.

The chemical modification of peptide chains after elongation would make it possible to quickly supply derivatives of already available parent peptide compounds. Although great effort has been devoted in recent years to develop chemical transformations that can achieve this feature, most existing methods depend on conventional functional-group transformations (FGTs) and require the presence of reactive functional groups in the side chains19); while they are expected to have high specificity, the applicable substrates and situations are severely limited.
Against this background, we devised the “N-chloropeptide strategy,” which involves N-chlorination of the amides in the peptide main chain, as a new method to achieve the chemical modification of peptide compounds20) (Chart 11). Secondary amides are ubiquitous in peptides, and some previous reports have shown that N-chlorination of conventional amides can be achieved via simple treatment with an electrophilic chlorinating agent,21,22) in contrast to the harsh conditions required by most N-functionalization reactions. We envisioned that N-chloropeptides, which had not previously been synthesized, could potentially be easily prepared from a native peptide and converted to a wide variety of non-proteinogenic AA structures through the following transformation using the reactive N–Cl bond of N-chloroamide. This approach does not require the preinstallation of any specific AAs. We expected that this feature would enable not only shorter synthetic routes to peptide fragments that contain unusual AAs but also the late-stage modification of peptide molecules.

We started with the investigation of the N-chlorination of simple dipeptide 128 as a model substrate (Table 6). Initially, dipeptide 128 was treated with t-butyl hypochlorite and trichloroisocyanuric acid (TCCA), which had already been used in the N-chlorination of simple amides.21,22) While t-butylhypochlorite (tBuOCl) did not modify peptide 128, the reaction using TCCA in MeCN for 3 h provided the desired N-chloropeptide 129 in 15% yield (entries 1 and 2). Notably, 129 can be purified by column chromatography on silica gel. Isolated 129 is stable under atmospheric conditions and can be stored in a refrigerator for over a year without any tangible degradation. While the conversion of peptide 128 was improved by extending the reaction time to 24 h, the reaction was still quite slow at room temperature (entry 3) and further screening of other commercially available chlorinating agents did not improve the conversion or yield relative to TCCA. To improve the reactivity, we turned from neutral chlorinating agents to in situ-generated cationic species, which typically have stronger electrophilic properties. We screened various nucleophilic catalysts in the presence of tBuOCl and found that the addition of a catalytic amount of pyridine, 1,4-diazabicyclo [2.2.2]octane (DABCO), or 1-azabicyclo [2.2.2]octane (ABCO, quinuclidine) promoted the N-chlorination reaction effectively (entries 4–6), and that among these, ABCO was the most effective to afford N-chloropeptide 129 in 92% yield (entry 6). ABCO exhibits extraordinarily high catalytic activity, that is, 0.1 mol% of ABCO is sufficient to achieve full conversion. In other words, the reaction is slow but reaches completion upon extending the reaction time (entry 7). Moreover, a mixed solvent (MeCN : H2O = 1 : 1, v/v) can be used for this transformation, which would be advantageous for the chlorination of longer hydrophilic peptide chains (entry 8).
 |
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Cl source | Time (h) |
Conversion (%) |
Yield (%) |
| 1 | None | tBuOCl | 3 | <5 | 0 |
| 2 | None | TCCA | 3 | 19 | 15 |
| 3 | None | TCCA | 24 | 75 | 68 |
| 4 | Pyridine | tBuOCl | 1 | 19 | 20 |
| 5 | DABCO | tBuOCl | 1 | 47 | 49 |
| 6 | ABCO | tBuOCl | 1 | >95 | 92 (92) |
| 7a) | ABCO | tBuOCl | 12 | >95 | 98 |
| 8b) | ABCO | tBuOCl | 0.5 | >95 | 100 |
Conversion and yield values were determined based on 1H-NMR spectroscopy using dimethyl terephthalate as the internal standard. Isolated yields are given in parentheses. a) The reaction was performed using 0.1 mol% of ABCO in toluene (0.5 M). b) MeCN/H2O (1 : 1, v/v) was used as the solvent.
Dehydro-AAs (ΔAAs) are a unique class of non-proteinogenic AAs that can be found in a variety of bioactive natural products. Conventional methods for the construction of ΔAAs require the preinstallation of specific AAs.23) We envisioned that N-chloropeptide 129 could be easily converted to the corresponding ΔAA-containing peptides via β-elimination in the presence of base, based on the X-ray structure of N-chloropeptide 129, in which N–Cl and C–H bonds are arranged in an almost antiperiplanar fashion (Table 6). Indeed, treatment with bicyclic amines such as DABCO and ABCO afforded the desired peptide (130) in excellent yield20) (Chart 12). In addition, we found that N-chlorination and β-elimination can be applied to a one-pot sequence even at the 5-mmol scale, and the desired peptide (130) was successfully obtained in 92% yield.

The optimized protocol enables the rapid construction of a wide variety of ΔAA residues (Table 7a). Bulky ΔAAs with β-substituents were easily introduced into dipeptides, and the desired compounds (131–134) were obtained in excellent yield. Aromatic-ring-conjugated double bonds were successfully installed into peptides; remarkably, electron-rich aromatic rings such as phenol and indole were well tolerated under the oxidative conditions (135–137). Asp, Asn, and O-methyl-Ser residues were also successfully converted into the corresponding ΔAA residues (138–140). Conventional methods based on β-elimination require specific β-functionalized AAs, whose preparation usually involves several steps. In contrast, using our method, a variety of ΔAA residues were easily constructed by simply changing the starting AA. In addition, we confirmed that N-Boc-protected peptides could also be converted to the corresponding ΔAA-containing peptide; while the N-chlorination on t-butyl carbamate competed with the N-chlorination on the amide, the dichlorination proceeded smoothly in the presence of an excess of tBuOCl, and treatment of the dichlorinated peptides with DABCO selectively promoted β-elimination on N-chloroamide. Subsequent reduction of the remaining N-chlorocarbamate with aqueous Na2SO3 afforded the desired ΔAA-containing peptides (141, 142) in good yield.
 |
 |
 |
Isolated yields are shown. a) The N-chlorination was performed at 0°C. b) Saturated aqueous Na2SO3 was used after the reaction.
Subsequently, the late-stage installation of a ΔAA motif into a macrocyclic peptide was examined as a very powerful demonstration of this method (Table 7b). Cyclosporin A, a calcineurin inhibitor used as an immunosuppressant medication, is a macrocyclic undecapeptide mostly consisting of aliphatic side chains. Cyclosporin analogue 143 was dichlorinated under the aforementioned conditions, and subsequent treatment with ABCO provided mono-dehydrogenated product 145 as a single regioisomer in moderate yield. This is the first example to achieve a late-stage introduction of a ΔAA motif into macrocyclic peptides; this result demonstrates that the N-chloropeptide strategy represents a powerful approach to modify aliphatic peptides.
4.3. C(sp3)–H Chlorination of Oligopeptides through γ- and δ-Selective Hydrogen-Atom Transfer (HAT)Chlorine-containing compounds are frequently found as naturally occurring analogues of biomolecules, and peptides containing C–Cl bonds in aliphatic side chains are also attractive molecules considering their biosynthesis and bioactive properties. In addition, the polar C(sp3)–Cl bonds are a reactive functional group for subsequent transformations. However, previously, only conventional FGTs had been available to install a chlorine atom into peptide molecules; therefore, a new method to efficiently provide a wide variety of chlorine-containing peptides was in high demand (Chart 13a). Another straightforward synthetic approach toward alkyl chlorides is C(sp3)–H functionalization, and several methods applicable to the late-stage chlorination of complex molecules have recently been reported (Chart 13b). However, the previous methods, in which a highly electrophilic species abstracts intermolecular C(sp3)–H bonds, predominantly engage tertiary C–H bonds based on the stability of the generated radical. In addition, the reaction of the peptide was limited to a single substrate, probably due to the low reactivity of aliphatic C–H bonds in the peptide side chains.24,25)

Against this background, we envisioned that the N-chloropeptide strategy could potentially be suitable for C(sp3)–H chlorination via a 1,5-HAT reaction of the amidyl radical, and that appropriate conditions could potentially enable the synthesis of a wide variety of chlorinated peptides including primary, secondary, and tertiary C–Cl bonds in a site-selective and predictable manner, taking advantage of the intramolecular HAT process. Based on this concept, we explored the 1,5-HAT reaction using N-chloropeptides and found that copper-catalyzed conditions followed by 1,5-HAT successfully provide the desired γ-chlorinated products (146, 147) in 80 and 91% yield from native Me- or tBu-protected peptides without the need to isolate the N-chloropeptide intermediate26) (Table 8). The presence of a bulky substituent at the C-terminus did not affect the efficiency of the C(sp3)–H chlorination reaction (148), while secondary and tertiary C–H bonds at the γ-position provided the corresponding chlorinated peptides (149, 150) in 77 and 88% yield, respectively. This approach was also effective for a Boc-protected peptide under modified conditions using TCCA in the N-chlorination step (151). A C–H bond at the C-terminus from the chlorinated amide was also accessible, and the δ-position of the isoleucine (Ile) motif was successfully chlorinated; this reaction allowed the gram-scale synthesis of chlorinated peptide 152 without any precautions, affording 1.60 g of the desired product. Moreover, bulky unusual AA residues were suitable for this transformation, affording the corresponding γ-chlorinated analogues in excellent yield (153–155). Polar AA side chains were efficiently chlorinated despite the less-accessible sites in the intermolecular HAT process owing to the radical polar effect (156–160). Notably, an electron-rich aromatic ring tolerated the electrophilic N-chlorination and copper-catalyzed 1,5-HAT conditions, providing the corresponding γ-chlorinated peptides (161, 162) in excellent yield.
 |
 |
Isolated yields. a) TCCA was used instead of ABCO and ‘BuOCI and the N-chloropeptide intermediate were purified to remove the byproducts derived from TCCA.
When the reaction was examined with substrates that contain multiple reactive C–H bonds, we found that tertiary C–H bonds react preferentially compared to primary C–H bonds (163). Interestingly, when a dipeptide consisting of Val and Ala was subjected to these reaction conditions, the reaction proceeded selectively at 1 methyl group of the Val residue (164). More surprisingly, when the C–H chlorination was repeated using 164 one or two additional times, the dichlorinated and trichlorinated products 165 and 166 were successfully obtained as single diastereomers, respectively, probably due to the limitation of the C–C bond rotation between the α- and β-carbon with the introduction of the bulky phthaloyl-protecting group.
Aquimarins are natural peptides with antimicrobial activity that were first isolated and structurally determined in 2022; 4 of these derivatives were found to contain chlorinated Ile residues at the γ-position27) (Chart 14). Although the synthesis of chlorine-free analogues has already been achieved by SPPS, it is not easy to synthesize chlorinated Ile residues containing 3 chiral centers, and the configuration of the chlorinated Ile residues has not yet been clarified. We envisioned that our new method to introduce chlorine atoms to peptide side chains could potentially enable the rapid preparation and determination of the stereo-configuration of chlorinated Ile residues. Indeed, we succeeded in obtaining a γ-chlorinated dipeptide consisting of Boc-protected Ile and Ala by sequential N-chlorination and 1,5-HAT conditions. Fortunately, it was possible to separate the 2 diastereomers 168a and 168b by column chromatography, and the configuration of dipeptide 168b was determined unequivocally by single-crystal X-ray diffraction analysis. After deprotection of the Boc groups of diastereomers 168a and 168b, the N-terminal dipeptide fragment of aquimarin A was successfully synthesized by condensation with (R)-3-hydroxyisocaproic acid. Furthermore, comparison of the 1H- and 13C-NMR spectra of the obtained compounds 169a and 169b strongly suggested that the stereoconfiguration of the chlorine-substituted carbon of the chlorinated Ile residue in natural products containing aquimarin A is the R configuration.

Finally, we examined the further derivatization of the side-chain-chlorinated peptides synthesized thus far (Chart 15). The N-terminal phthaloyl group of dipeptide 147 was easily deprotected with hydrazine, and a subsequent condensation reaction with a Boc-protected AA yielded the elongated tripeptide 170 in high yield. The C-terminal t-butyl ester was also easily removed, and tetrapeptide 171 was successfully obtained without epimerization by fragment condensation of the dipeptide. Notably, the C–Cl bond introduced into the side chain did not react at all in these series of deprotection and condensation reactions, indicating that the side-chain-chlorinated AA residue is a stable structure that can be applied in normal peptide-elongation operations. On the other hand, the C–Cl bond can be converted to other functional groups under appropriate conditions. In fact, γ-functionalized peptides 172–174 were obtained in good yield through the action of nucleophiles such as azide and iodide anions or thiophenols on the side-chain-chlorinated tripeptide 170. These results show that side-chain-chlorinated peptides are extremely useful building blocks for the preparation of unusual peptide structures.

In this review, 3 unconventional synthetic approaches toward unusual peptide derivatives developed in our group are described. The external-oxidant-mediated decarboxylative condensation of α-ketoacid represents a new option for peptide elongation as well as the chemoselective transformation of complex molecules. An organocatalytic asymmetric Mannich-type addition provides a wide variety of chiral β-amino-α-ketoacid equivalents, which are difficult to access using the conventional chiral-pool approach. Furthermore, we are also currently developing the “N-chloropeptide strategy” for the chemical modification of peptides and have already succeeded in not only the preparation of a wide range of unusual peptide structures but also the late-stage modification of macrocyclic peptides. These unique synthetic approaches are expected to contribute to peptide-based drug discovery.
I would like to thank all my collaborators, and especially Prof. Yoshiji Takemoto for his support and fruitful advice. This research was partially supported by JSPS KAKENHI grants JP18K14865, JP20K15954, and JP22H02743. I would also like to gratefully acknowledge the Takeda Science Foundation, the Astellas Foundation for Research on Metabolic Disorders, the Chugai Foundation for Innovative Drug Discovery Science, and the Uehara Memorial Foundation. I am moreover grateful to Dr. Hiroyasu Sato (Rigaku) for his assistance with the analysis of X-ray crystallography data.
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2024 Pharmaceutical Society of Japan Award for Young Scientists.