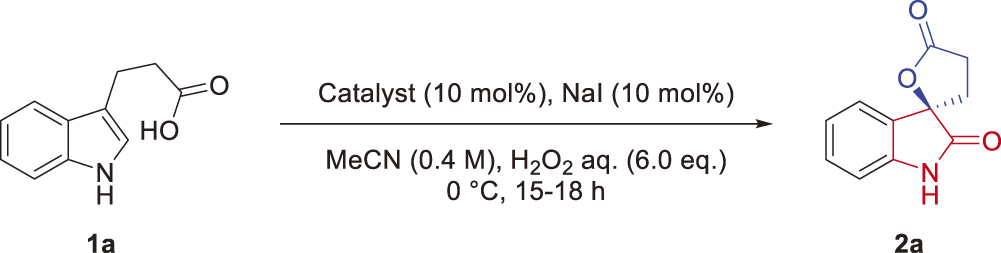Experimental
General Methods
1H-NMR and 13C-NMR spectra were recorded on a Bruker Avance III Nanobay 400 MHz spectrometer (400 MHz for 1H-NMR, 100 MHz for 13C-NMR). The chemical shifts were expressed in ppm downfield from tetramethylsilane (δ=0.00) as an internal standard and CDCl3 (δ=77.16), CD3OD (δ=49.00). Mass spectra were recorded by an electrospray ionization-time of flight (ESI-TOF) mass spectrometer (Xevo G2-XS QTof). Melting points were obtained with Yanaco MP-J3. For TLC analyses, Merck precoated TLC plates (silica gel 60F254) were used. Flash column chromatography was performed on neutral silica gel (Kanto Silica gel 60N, 40–50µm) HPLC analysis was conducted using Shimadzu LC-20AT coupled diode array-detector SPD-M20A and chiral column of DAICEL ChiralPak IA (4.6 × 250 mm), DAICEL ChiralPak IC (4.6 × 250 mm), DAICEL ChiralPak IG (4.6 × 250 mm). 2a–o are known compounds that exhibited spectroscopic data identical to those reported in the literature.28,30)
A typical procedure of the asymmetric oxidative cyclization of 1a using organocatalyst C5 is as follows: To a stirred solution of 3-(1H-indol-3-yl) propanoic acid (1a, 37.8 mg, 0.200 mmol), NIS (4.50 mg, 20.0 μmol) and organocatalyst C5 (12.6 mg, 20.0 μmol) in MeCN (0.5 mL), and 35% H2O2(aq.) (103µL, 1.20 mol) were added at 0°C. After stirring at 0°C for 16h, the reaction mixture was diluted with EtOAc (15 mL) and successively washed with 10% Na2S2O3(aq.) (15 mL), sat. NaHCO3(aq.) (15 mL) and brine. The organic layer was dried over Na2SO4, filtered, and evaporated. The obtained residue was purified by flash column chromatography on silica gel with a 5 : 1–3 : 1 mixture of hexane and EtOAc to afford (R)-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2a) (26.8 mg, 0.132 mmol, 66%, 75% ee) as a white solid. 1H-NMR (CDCl3, 400 MHz) δ: 8.57 (brs, 1H), 7.36–7.32 (m, 2H), 7.12 (t, J=7.6 Hz, 1H), 6.94 (d, J=7.9 Hz, 1H), 3.18 (dt, J=17.8, 10.1 Hz, 1H), 2.79 (ddd, J=17.7, 9.6, 2.9Hz, 1H), 2.63 (ddd, J = 13.2, 9.8, 3.0Hz, 1H), 2.48 (dt, J = 13.3, 10.2 Hz, 1H); 13C-NMR (CDCl3, 100 MHz) δ: 176.8, 176.5, 141.2, 131.3, 126.8, 124.6, 123.7, 111.1, 82.9, 31.4, 28.3; high resolution (HR)MS (ESI-TOF) m/z: [M+Na]+ Calcd for C11H9NO3Na+, 226.0475; Found, 226.0474. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 80/20, 1.0 mL/min), λ = 254 nm: tminor = 25.2 min, tmajor = 28.0 min. [α]D26 = −9.6 (c 1.0, CH2Cl2).
(R)-4′-Methyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2b)
1H-NMR (CDCl3, 400 MHz) δ: 8.23 (brs, 1H), 7.80 (t, J = 7.8 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.75 (d, J = 7.7 Hz, 1H), 3.22 (dt, J = 17.6, 10.6 Hz, 1H), 2.77 (ddd, J = 17.6, 9.8, 2.5 Hz, 1H), 2.69–2.52 (m, 2H), 2.34 (s, 3H); 13C-NMR (CD3OD, 100 MHz) δ: 177.4, 177.0, 142.0, 136.2, 130.6, 125.0, 124.0, 108.0, 83.9, 28.3, 27.3, 16.0. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: tminor = 14.3 min, tmajor = 17.9 min. [α]D27 = −35.4 (c 1.0, CH2Cl2).
(R)-5′-Methyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2c)
1H-NMR (CDCl3, 400 MHz) δ: 8.29 (brs, 1H), 7.16 (s, 1H), 7.13 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 7.9 Hz, 1H), 3.17 (ddd, J = 17.7, 10.5, 9.8 Hz, 1H), 2.78 (ddd, J = 17.6, 9.6, 3.1 Hz, 1H), 2.65–2.58 (m, 1H), 2.50–2.42 (m, 1H), 2.33 (s, 3H); 13C-NMR (CD3OD, 100 MHz) δ: 178.8, 178.2, 140.8, 134.2, 132.5, 128.5, 126.3, 111.4, 84.7, 32.0, 29.1, 21.0. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 80/20, 1.0 mL/min), λ = 254 nm: tminor = 26.3 min, tmajor = 32.0 min. [α]D27 = −14.0 (c 1.0, CH2Cl2).
(R)-6′-Methyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2d)
1H-NMR (CDCl3, 400 MHz) δ: 8.48 (brs, 1H), 7.22 (d, J = 7.7 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 3.17 (dt, J = 17.8, 10.4 Hz, 1H), 2.77 (ddd, J = 17.6, 9.6, 3.0 Hz, 1H), 2.63–2.57 (m, 1H), 2.49–2.41 (m, 1H), 2.36 (s, 3H); 13C-NMR (CD3OD, 100 MHz) δ: 177.4, 177.0, 142.1, 141.6, 124.13, 124.08, 123.4, 111.0, 83.3, 30.5, 27.8, 20.5. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 85/15, 1.0 mL/min), λ = 254 nm: tminor = 48.5 min, tmajor = 51.8 min. [α]D27 = −4.6 (c 0.85, CH2Cl2).
(R)-7′-Methyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2e)
1H-NMR (CDCl3, 400 MHz) δ: 8.26 (brs, 1H), 7.17 (t, J = 7.3 Hz, 2H), 7.04 (t, J = 7.6 Hz, 1H), 3.17 (ddd, J = 17.6, 10.6, 9.8 Hz, 1H), 2.78 (ddd, J = 17.6, 9.6, 3.0 Hz, 1H), 2.65–2.59 (m, 1H), 2.50–2.44 (m, 1H), 2.27 (s, 3H); 13C-NMR (CD3OD, 100 MHz) δ: 177.4, 177.1, 140.4, 132.1, 126.7, 122.9, 121.6, 120.2, 83.5, 30.7, 27.7, 15.1. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 80/20, 1.0 mL/min), λ = 254 nm: tminor = 29.1 min, tmajor = 37.3 min. [α]D26 = −7.2 (c 1.0, CH2Cl2).
(R)-4′-Methoxy-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2f)
1H-NMR (CD3OD, 400 MHz) δ: 7.31 (t, J = 8.2 Hz, 1H), 6.73 (d, J = 8.5 Hz, 1H), 6.55 (dd, J = 7.8, 0.6 Hz, 1H), 3.87 (s, 3H), 2.93–2.88 (m, 2H), 2.65–2.58 (m, 1H), 2.50 (dt, J = 13.4, 8.6 Hz, 1H); 13C-NMR (CD3OD, 100 MHz) δ: 177.8, 176.5, 157.0, 143.0, 132.4, 113.4, 105.9, 103.5, 83.3, 54.7, 28.0, 27.6. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 80/20, 1.0 mL/min), λ = 254 nm: tminor = 45.1 min, tmajor = 68.4 min. [α]D25 = 4.8 (c 0.85, CH2Cl2).
(R)-5′-Methoxy-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2g)
1H-NMR (CD3OD, 400 MHz) δ: 7.10 (d, J = 2.5 Hz, 1H), 6.91 (dd, J = 8.5, 2.6 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H), 3.78 (s, 3H), 3.08–2.99 (m, 1H), 2.81 (ddd, J = 17.7, 7.9, 5.0 Hz, 1H), 2.56–2.51 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 177.4, 176.8, 156.6, 134.9, 128.2, 115.8, 110.9, 83.6, 54.9, 30.6, 27.7. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 80/20, 1.0 mL/min), λ = 254 nm: tminor = 35.9 min, tmajor = 39.2 min. [α]D25 = −12.6 (c 1.0, CH2Cl2).
(R)-5′-Fluoro-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2h)
1H-NMR (CD3OD, 400 MHz) δ: 7.31 (dd, J = 7.9, 2.6 Hz, 1H), 7.10 (td, J = 9.0, 2.7 Hz, 1H), 6.90 (dd, J = 8.6, 4.2 Hz, 1H), 3.08–2.98 (m, 1H), 2.81 (ddd, J = 17.7, 9.0, 4.0 Hz, 1H), 2.61–2.48 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 177.0, 176.7, 159.4 (q, JC-F = 240.5 Hz), 138.0, 128.7 (q, JC-F = 8.1 Hz), 117.1 (q, JC-F = 23.8 Hz), 112.2 (q, JC-F = 25.6 Hz), 111.3 (q, JC-F = 8.0 Hz), 83.1, 30.5, 27.5. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: tminor = 12.5 min, tmajor = 10.4 min. [α]D25 = −7.2 (c 0.63, CH2Cl2).
(R)-6′-Fluoro-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2i)
1H-NMR (CD3OD, 400 MHz) δ: 7.46 (dd, J = 8.3, 5.3 Hz, 1H), 6.81 (ddd, J = 9.7, 8.3, 2.2 Hz, 1H), 6.69 (dd, J = 8.9, 2.3 Hz, 1H), 3.08–2.99 (m, 1H), 2.80 (ddd, J = 17.7, 8.6, 4.3 Hz, 1H), 2.60–2.48 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 177.1, 176.9, 164.6 (q, JC-F = 247.1 Hz), 144.0 (q, JC-F = 12.4 Hz), 126.2 (q, JC-F = 10.4 Hz), 122.8 (q, JC-F = 3.0 Hz), 109.0 (q, JC-F = 23.2 Hz), 98.6 (q, JC-F = 27.8 Hz), 82.6, 30.4, 27.7. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: tminor = 9.4 min, tmajor = 10.8 min. [α]D26 = −51.0 (c 0.8, CH2Cl2).
(R)-7′-Fluoro-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2j)
1H-NMR (CD3OD, 400 MHz) δ: 7.30–7.28 (m, 1H), 7.20–7.07 (m, 2H), 3.08–2.99 (m, 1H), 3.04 (dt, J = 17.7, 9.9 Hz, 1H), 2.82 (ddd, J = 17.7, 9.3, 3.8 Hz, 1H), 2.63–2.49 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 177.0, 176.2, 148.3, 145.9, 129.9 (q, JC-F = 3.5 Hz), 123.8 (q, JC-F = 6.0 Hz), 120.2 (q, JC-F = 3.5 Hz), 117.6 (q, JC-F = 17.6 Hz), 82.9, 30.6, 27.5. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: tminor = 11.6 min, tmajor = 27.7 min. [α]D26 = −5.1 (c 0.6, CH2Cl2).
(R)-5′-Chloro-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2k)
1H-NMR (CD3OD, 400 MHz) δ: 7.52–7.51 (m, 1H), 7.36–7.33 (m, 1H), 6.92–6.89 (m, 1H), 3.02 (dt, J = 17.7, 10.0 Hz, 1H), 2.81 (ddd, J = 17.7, 8.7, 4.3 Hz, 1H), 2.60–2.49 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 177.0, 176.4, 140.8, 130.7, 128.9, 128.1, 124.9, 111.5, 82.8, 30.4, 27.5. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IG, Hexane/i-PrOH = 90/10, 0.5 mL/min), λ = 254 nm: tminor = 43.9 min, tmajor = 46.2 min. [α]D26 = −15.2 (c 1.0, CH2Cl2).
(R)-4′-Bromo-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2l)
1H-NMR (CD3OD, 400 MHz) δ: 7.28–7.22 (m, 2H), 6.92 (dd, J = 7.0, 1.7 Hz, 1H), 3.01–2.99 (m, 1H), 2.92–2.79 (m, 2H), 2.57–2.49 (m, 1H); 13C-NMR (CD3OD, 100 MHz) δ: 177.1, 176.1, 144.2, 132.4, 126.5, 125.4, 119.2, 109.7, 83.5, 27.4, 27.0. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: tminor = 11.1 min, tmajor = 15.7 min. [α]D27 = −4.5 (c 1.0, CH2Cl2).
(R)-5′-Bromo-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2m)
1H-NMR (CD3OD, 400 MHz) δ: 7.65–7.64 (m, 1H), 7.51–7.47 (m, 1H), 6.86 (d, J = 8.3 Hz, 1H), 3.02 (dt, J = 17.4, 10.0 Hz, 1H), 2.81 (ddd, J = 17.7, 8.7, 4.3 Hz, 1H), 2.57–2.51 (m, 2H); 13C-NMR (CD3OD, 100 MHz) δ: 178.4, 177.7, 142.7, 135.0, 130.7, 129.1, 116.4, 113.4, 84.1, 31.8, 28.9. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IA, Hexane/i-PrOH = 90/10, 1.0 mL/min), λ = 254 nm: tminor = 20.7 min, tmajor = 22.8 min. [α]D27 = −11.0 (c 1.0, CH2Cl2).
1′-Methyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2n)
1H-NMR (CDCl3, 400 MHz) δ: 7.43–7.35 (m, 2H), 7.16–7.12 (m, 1H), 6.87 (d, J = 7.8 Hz, 1H), 3.25–3.16 (m, 1H), 3.20 (s, 3H), 2.77 (ddd, J = 17.6, 9.5, 3.1 Hz, 1H), 2.58 (ddd, J = 17.6, 9.5, 3.1 Hz, 1H), 2.46 (ddd, J = 13.3, 10.6, 9.5 Hz, 1H). 13C-NMR (CD3OD, 100 MHz) δ: 178.6, 176.2, 145.2, 132.4, 127.9, 125.4, 124.8, 110.4, 84.2, 31.9, 29.1, 26.6. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: t = 43.1 min, t = 48.3 min.
1′-Benzyl-3,4-dihydro-5H-spiro[furan-2,3′-indoline]-2′,5-dione (2o)
1H-NMR (CDCl3, 400 MHz) δ: 7.37–7.27 (m, 7H), 7.10 (t, J = 7.1 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H), 4.88 (s, 2H), 3.25 (ddd, J = 17.6, 10.6, 9.8 Hz, 1H), 2.84–2.76 (m, 1H), 2.63 (ddd, J = 13.2, 9.8, 3.1 Hz, 1H), 2.50 (ddd, J = 13.3, 10.7, 9.6 Hz, 1H). 13C-NMR (CDCl3, 100 MHz) δ: 176.1, 174.4, 143.0, 134.9, 131.1, 129.0, 128.0, 127.2, 126.4, 124.3, 123.6, 109.9, 82.3, 43.9, 31.5, 28.3. The enantiomeric excess of the product was determined by chiral HPLC analysis using a DAICEL ChiralPak IC, Hexane/i-PrOH = 70/30, 1.0 mL/min), λ = 254 nm: t = 23.7 min, t = 29.6 min.