Abstract
Primary macronodular adrenal hyperplasia (PMAH), also known in the past as bilateral macronodular adrenalhyperplasia or adrenocorticotropin (ACTH)-independent macronodular adrenal hyperplasia, is a rare type of Cushing’s syndrome (CS) and is associated with bilateralenlargement of the adrenal glands. It accounts for <1% of all endogenous cases of CS. In order toidentify the pathogenic mutations in the causative gene of (AIMAH pedigrees, Whole-genome sequencing of three patients in family I was used to retrieve candidate causative genes. Meanwhile, the causative gene was identified by Sanger sequencing from the two pedigrees. Sequencing of ARMC5 exons of three patients was carried out to identify somatic mutations. Moreover, haploid clone of one tumor DNA sample was conducted. ARMC5 was the causative gene of two pedigrees confirmed by whole-genome sequencing (WGA) and Sanger sequencing. The variant sites of the two families were c.C943T (p.R315W) and c.C1960T (p.R654X), respectively. Autosomal dominant inheritance of AIMAH was confirmed by genotypes of one family member. Several somatic mutations were discovered in tumor DNA samples. In addition, haploid clone of tumor DNA was confirmed by germline mutation and somaticmutation, which suggested the pathogenic mechanism of “two-hit-model.” ARMC5 was the causative gene of AIMAH pedigrees. This AIMAH in this study presented autosomal dominant inheritance, fitting to Mendelian inheritance law. However, the pathogenic mode of this disease showed as compound heterozygote.
ACTH INDEPENDENT MACRONODULAR ADRENAL HYPERPLASIA (AIMAH, OMIM #615954), also known as primary bilateral macronodular adrenal hyperplasia [1], is a rare disorder characterized by bilateral macronodular hyperplasia of the adrenal glands with increased cortisol production. Patients typically present in the fifth and sixth decades of life with obvious clinical manifestations of moon face, sanguineous temperament, skin thinning, buffalo hump and thickened supraclavicular fat pad. Bilateral adrenalectomy followed by replacement therapy with prednisone is usually adopted for disease treatment. In the majority of cases AIMAH appears to be sporadic. Several reports of familial clustering have been published with an autosomal dominant pattern of transmission [2]. AIMAH represents less than 1% of cases of endogenous Cushing’s syndrome (CS); however, as 10% of incidentally found adrenal lesions are bilateral, AIMAH with subclinical cortisol secretion is being increasingly recognized [3]. The most common clinical presentation is subclinical, followed by clinical CS. Patients with AIMAH respond to ACTH with a substantial increase in cortisol, which helps to distinguish it from other causes of bilaterally enlarged adrenals such as metastatic or infiltrative diseases [4]. The hormone secretion in AIMAH results from an increase in the number of adrenocortical cells rather than an augmented synthesis per cell. There is a relatively inefficient hormonal synthesis in AIMAH, with decreased expression of several steroidogenic enzymes and higher levels of certain precursors such as plasma 17-hydroxyprogesterone or urinary 17-hydroxycorticosteroids [5]. This was confirmed by various immunohistochemicalsteroidogenic enzyme studies [6]. This inefficient steroidogenesis may explain the incongruity of some subclinical cortisol hypersecretion despite massive adrenal enlargement.
Previously, periodic clinical screening using biochemical and radiological investigations were the only means by which potentially affected individuals could be identified. However, this is labor intensive and subjects many unaffected individuals to invasive investigations including radiation exposure from adrenal imaging. Genetic testing of kindreds after diagnosis of an individual affected by AIMAH may lead to earlier diagnosis by enabling targeted screening of those individuals harboring the genetic mutation andwhowould therefore benefit from periodic clinical screening while sparing those without the causative genetic mutation. The genetic basis of a majority (44%–55%) of cases of AIMAH studied has been elucidated recently and found to be due to germline mutations in armadillo repeat containing 5 (ARMC5) [7, 8]. Furthermore, damagingARMC5 mutations were associated with a more severe CS phenotype [8]. Somatic second hit-mutations of ARMC5 were present in adrenal tumors, suggesting ARMC5 functions as a tumor suppressor gene [7]. In vitro, ARMC5 mutations resulted in decreased steroidogenesis and altered cell survival, which explains the observations of inefficient steroidogenesisand hyperplasia in AIMAH [9]. ARMC5 makes part of a large family of proteins, characterized by the presence of tandem repeats of an amino acid motif. This family is highly conserved through evolution in eukaryotic organisms (from yeast to human) [10]. Previous studies found that beyond a certain common event, such as ectopic expression of GIP receptors or germline ARMC5 mutations in diffuse hyperplasia, several somatic genetic events occurred in the different macronodules. Thus, the somatic ARMC5 mutation or another hit may contribute to the progression of macronodules. However the function of ARMC5 remains unknown as do the mechanisms of the inactivating mutations of ARMC5 involved in development of adrenal macronodular hyperplasia [11]. Interestingly, six out of 11 (54%) first-degree relatives of seven probands carried a germline ARMC5 mutation; five of these showed at least one nodule on computed tomography (CT) scans, coupled with increased cortisol secretion in three cases [12]. One elderly patient (76 years of age) harboring a germline mutation did not show any abnormal hormonal results and showed a normal adrenal CT scan. It is possible that this patient did not have a second-hit ARMC5 molecular event in the adrenal gland, indicative of incomplete penetrance of AIMAH [12]. The bilateral nature of AIMAH, on the other hand, strongly suggests the involvement of genetic factors for disease development. Several causal genes of Cushing’s syndrome had been identified recently, including GNAS1 for McCune-Albright syndrome [13] AR1A for Carney Complex [14] PDE11A [15], PDE8B [16], MEN1 [17], and PRKACA[18-20]. Importantly, ARMC5 was revealed as a recurrent driver mutation in AIMAH by analysis of 33 sporadic patients [7]. It suggested that ARMC5 functions as a tumor-suppression gene following the two-hit model, where a primary germ-line mutation in one allele and a somatic secondary inactivation of the second allele resulted in adrenal hyperplasia. High frequency of mutation occurrence (18 out of 33 patients, 55%) implied a pivotal role of ARMC5 in disease etiology. While both adrenal nodules and blood samples were analyzed in parallel for ARMC5 mutations. However, origin of the germ-line mutations was not elucidated by Assié et al. for the sporadic nature of these patients [7]. The purpose of this investigation was to search for novel ARMC5 mutations in such patients in China.
In this study, we take advantage of whole genome sequencing technology to analyze the germ-line causal mutation(s) in two affected families. By filtering those shared rare variants among three patients from family I against dbSNP139 and the 1,000 Genomes Project database, two novel mutations of ARMC5 (c.C943T (p.R315W) and c.C1960T (p.R654X)) were revealed, where the two-hit model explained the adrenal hyperplasia in more than one patient. The novel germline ARMC5 mutations identified in this study extend the spectrum of ARMC5 mutations in AIMAH.
Method and Materials
Clinical samples and Illumina sequencing
All methods were performed in accordance with the relevant guidelines and regulations. Two affected AIMAH families were enrolled for this study with written informed consent forms of each participant. This research was approved by the ethics committee of The PLA General Hospital (KLS32013-107-01). Of the first family, there were twelve members who participated in this study. Six members had been identified as AIMAH patients, including II3, II5, II9, II11, II13 and II15. It was worthy noticed that II9 and II11 owned the typically clinical features of CS. Meanwhile, II3, II5, II13 and II15 had the sub-clinical features of CS (SCS). Of the second family, there were eighteen members who participated in this study. Three members had been identified as AIMAH patients, including II3, II9 and III8. It was worthy noticed that II3 owned the typically clinical features of CS. Meanwhile, II9 and III8 had the sub-clinical features of CS (SCS). Moreover, the computed tomographic (CT) imaging technique and H&E staining methods were employed to identify the characteristics of nodular hyperplasia. For CT analysis, the images were reviewed by two experienced observers, who arrived at a consensus. Imaging characteristics were recorded. For H&E staining, allsurgical specimens were fixed in 10% buffered formaldehyde and routinely processed for histologic diagnosis.
Genomic DNA was extracted from whole blood samples of patients II4, II6 and II8 of family I with DNeasy Blood & Tissue Kit (Qiagen). Sequencing libraries of 400-bp insert size were constructed from 2 μg genomic DNA with TruSeq DNA Sample Preparation Kit (Illumina), which was then sequenced on the HiSeq 2500 platform (Supplementary Table 1). Sequencing depth for these samples were 6X, 25X, and 38X, respectively.
Reads mapping and variant analysis
Illumina sequencing data were mapped to the human reference genome (NCBI37/hg19) with BWA. PCR duplicates and low quality reads in each dataset were removed by SAMtools. After local re-alignment of the reads surrounding indels, GATK (version 2.3-9-ge5ebf34) was used for variant calling, where at least three reads were required to support each variant. Considering the low sequencing coverage of patient II4, shared variants between II6 and II8 were firstly called, which were then filtered against II4 by allowing missing data. After common variant filtering by dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) (Build 139), rare variant shared across the three sequenced patients were annotated by ANNOVAR. Functional effects of candidate variants were evaluated by SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
Candidate variant validation
Genomic DNA from whole blood samples and removed adrenal gland tissues of some patients under surgery were extracted, and PCR primers surrounding each focal variant were designed by Primer3. PCR products were then sequenced on ABI 3730xl for genotype validation. For haploid phasing of ARMC5 mutations, long PCR primers encompassing candidate variants were designed, and PCR products were ligated into pGEM-T Easy vector for E. coli transformation. Five colonies were then picked up for Sanger sequencing from T7/SP6 primers. For Sanger sequencing, Genomic DNA (2.5 ng/μL, 10 μL reactions) was amplified (35 cycles) in a GeneAmp 9700 thermal cycler using Top Taqmastermix kit (Qiagen) and 20 pmol of gene-specific primers. Resulting PCR amplicons were enzyme-purified with ExoSAP-IT (USB Corporation, Cleveland, OH). The purified amplicons were direct cycle sequenced in both directions with BigDye Terminator Ready Reaction Mix (v3.1) (Applied Biosystems, Grand Island, NY) containing M13 forward or reverse sequencing primers, then ethanol precipitated and detected by capillary electrophoresis on a 3130XL Genetic Analyzer running Sequence Analysis (v.6.0) software (Applied Biosystems) and Chromas (v2.23) software (Technelysium, Tewantin, Queensland, Australia).
Results
Clinical evaluation
In the past decade, two AIMAH affected families had been diagnosed in our hospital. For the first family, six out of twelvememebers were diagnosed with typical or sub-clinical AIMAH syndrome (Fig. 1). The clinical features of this affected family were summarized in Table 1. None of the cases were products of a consanguineous marriage and all affected patients had bilateral congenital cataracts. The proband (II9) of family I was a man patient aged 66 who had suffered 4 years hypertension, hyperglycemia and 8 months central obesity before diagnosis. Pituitarium MRI examination did not implicate any positive results, while MRI imaging of Adrenal gland revealed irregular nodular mass on both the left (4.45 cm × 3.18 cm × 2.80 cm) and the right (2.8 cm × 2.0 cm × 1.5 cm) gland. CT scanning of adrenal gland showed that there was nodular shadow with uniformity density on both adrenal glands (Fig. 2). Left and right adrenalectomies were carried out independently within three years, followed by replacement therapy with prednisone at the dosage of 10 mg/day, and his blood pressure fall to 120/70 mmHg without any manifestations of Cushing’s syndrome. Histology of the removed adrenal glands showed that the adrenal cortex was composed of nodules with different sizes, and there were no normal cortex and medulla structure among the nodules, and all different sizes of cells contained clear cytoplasm (Fig. 3). Patient II11 recovered from right adrenalectomy by replacement therapy.

Table 1
Clinical evaluation of two Chinese pedigrees with AIMAH
| First family |
Second family |
Pedigree
number |
Gender |
Age |
Phenotype |
Pedigree
number |
Gender |
Age |
Phenotype |
| II-3 |
Female |
72 |
SCS |
II-3 |
Male |
67 |
CS |
| II-5 |
Male |
71 |
SCS |
II-6 |
Female |
65 |
Normal |
| II-9 |
Male |
66 |
CS |
II-8 |
Male |
63 |
Normal |
| II-11 |
Female |
64 |
CS |
II-9 |
Female |
61 |
SCS |
| II-13 |
Male |
62 |
SCS |
II-11 |
Male |
56 |
Normal |
| II-15 |
Female |
55 |
SCS |
II-14 |
Male |
54 |
Normal |
| III-5 |
Male |
40 |
Normal |
III-1 |
Male |
50 |
Normal |
| III-15 |
Female |
37 |
Normal |
III-6 |
Female |
50 |
Normal |
| III-18 |
Male |
29 |
Normal |
III-8 |
Male |
47 |
SCS |
| III-19 |
Male |
34 |
Normal |
III-11 |
Male |
46 |
Normal |
| III-20 |
Male |
27 |
Normal |
III-12 |
Female |
45 |
Normal |
| III-21 |
Male |
32 |
Normal |
III-17 |
Female |
40 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-21 |
Female |
32 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-22 |
Male |
40 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-25 |
Male |
39 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-26 |
Male |
33 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-30 |
Female |
31 |
Normal |
| N/A |
N/A |
N/A |
N/A |
III-33 |
Female |
30 |
Normal |


The proband (II3) of the second family was a man patient aged 67 recovered from left adrenalectomy by replacement therapy (Fig. 4). Pituitarium MRI examination did not implicate any positive results, while MRI imaging of adrenal gland revealed irregular nodular mass on both the left (6 cm × 3.5 cm × 3 cm) and the right (5 cm × 3 cm × 3 cm) gland. CT scanning of adrenal gland showed that there was nodular shadow with uniformity density on both adrenal glands (Fig. 2). Left adrenalectomies were carried out independently within three years, followed by replacement therapy with prednisone at the dosage of 10 mg/day, and his blood pressure fall to 120/70 mmHg without any manifestations of Cushing’s syndrome.
Sequencing and variant detection of three patients
To identify the causal mutation of the affected AIMAH families, three patients from family I, including II4, II6 and II8, were selected for whole-genome sequencing with IlluminaHiSeq 2500 platform. The results indicated that 110.6 Gb data (II: 11), 72.6 Gb data (II: 9) and 24.6 Gb data (II: 5) were mapped to human reference genome (hg19), respectively. The mean coverage of three samples across the whole-Genome was over 90%. Meanwhile, the average sequencing depth from these samples was 38X, 25X, and 6X, respectively (Table 2). After low quality reads filtering, these sequencing data were first mapped onto human reference genome (NCBI38/HG19) for variant identification, and common variant were filtered by dbSNP 139 version (NCBI) (https://www.ncbi.nlm.nih.gov/projects/SNP/) and The 1,000 Genomes Project database (http://www.internationalgenome.org/). Eventually, 95 non-synonymous SNVs and 30 coding Indels shared by II8 and II6 were identified. Meanwhile, 44 nsSNVs and 7 Indels were also present in II4 owning the lowest coverage (Fig. 5, Supplementary Table 1 and Supplementary Table 2). Moreover, structural variations were also analyzed by Pindel software (Supplementary Table 3).
Table 2
Summary of genome sequencing and mapping results
| Samples |
Data (Gb) |
PCR (%) |
Mapping (%) |
Coverage (%) |
Depth (X) |
| II11 |
115.16 |
0.90% |
96.06% |
91.46 |
38 |
| II9 |
75.72 |
0.50% |
95.89% |
91.73 |
25 |
| II5 |
25.587 |
0.50% |
96.18% |
90.48 |
6 |

It is noteworthy that none of known genes for Cushing’s syndrome were involved for these rare variants. SIFT and PolyPhen-2 analysis showed that 35 of these nsSNVs with damaging effects on protein function (Supplementary Table 4). Heterozygous variants and frame-shift Indels were then validated by PCR and Sanger sequencing within the affected family (Supplementary Table 5). The results indicated that only one nsSNV of ARMC5 segregated with disease phenotype, where all diagnosed patients as well as two individuals of the third generation (III18 and III19) were mutation heterozygotes. Another seemingly co-segregation nsSNV within FOXD4L5 turned out to be paralog sequence variant, which had four identical copies in human genome [21]. Taken together, these results strongly implied the involvement of ARMC5 in the etiology of AIMAH.
Mutation screen of the affected adrenal glands
Revelation of individuals carrying ARMC5 mutation without clinical or sub-clinical disease phenotype raised the possibility of two-hit model for disease development, which can be corroborated by its later diagnosis between 40 and 60 years of age. To clarify the mutation pattern of ARMC5 for its pathogenicity, we designed primers for each of the six coding exons of ARMC5 (Supplementary Table 5). Adrenal nodule samples from right adrenalectomy of patients II8 and II6 were used for PCR and sequencing. Samples from the second family, including blood DNAs of two patients and one adrenal nodule sample, as well as four adrenal nodules removed from sporadic Cushing’s patients, were also used for mutation screening.
Totally 9 coding variants were identified in ARMC5 across these samples (Table 3). Interestingly, the right adrenal nodule of patient II11 in family I had a 22-nt deletion in the first exon, as well as the germ-line R315W mutation of the third exon. Long PCR primers were then designed to phase ARMC5 haplotype of patient II11 (Supplementary Table 4). Colony sequencing unambiguously suggested that R315W mutation and the 22-nt deletion located on different haplotype. In addition, the R315W mutation had become homozygous in one right nodule of patient II9. On the other hand, sequencing of the second family revealed the presence of pre-mature stop codon on the 5th exon of ARMC5 as the germline mutation, while nodules of patient II5 demonstrated a distinct haplotype with 1-nt deletion in the first exon. Taken together, these data implied that realization of the adrenal hyperplasia phenotype in AIMAH required loss-of-function of ARMC5 in a recessive manner, which agreed well with Knudson’s two-hit model of cancer development [22]. Sequencing of four sporadic nodule tissues revealed only one heterozygous F842Y substitution in one sample, suggested that ARMC5 might not be culprit for these Cushing’s patients.
Table 3
Summary of nine coding variants identified in ARMC5 across samples
|
|
5' UTR |
#1 exon |
intron |
#3 exon |
#4 exon |
intron |
#5 exon |
#6 exon |
3' UTR |
| Variant |
Chr16 position |
3145
9197 |
3145
9219 |
31459773-
31459794 |
3145
9803 |
3146
0057 |
3146
2218 |
3146
2490 |
3146
4572 |
3146
5137 |
3146
5945 |
3146
6559 |
3146
6603 |
3146
6606 |
3146
7062 |
| Genotype |
C/T |
T/C |
CCCGTCCC
AGGCAGGC
CCCGGC/– |
C/– |
A/G |
C/A |
†C/T |
G/A |
C/T |
§C/T |
C/A |
G/A |
T/A |
C/T |
| AA change |
— |
— |
frame-shift |
frame-shift |
— |
A224E |
R315W |
E517K |
— |
R654* |
P826P |
R841H |
F842Y |
— |
| Family I |
*II3 |
— |
Homo |
— |
— |
— |
— |
Het |
— |
Homo |
— |
— |
— |
— |
— |
| *II5 |
— |
Homo |
— |
— |
— |
— |
Het |
— |
Homo |
— |
— |
— |
— |
— |
| *II9 |
— |
Het |
— |
— |
— |
— |
Het |
— |
Het |
— |
— |
— |
— |
— |
| *II9 right nodule 1 |
— |
Het |
— |
— |
— |
Het |
Het |
— |
Het |
— |
— |
— |
— |
— |
| *II9 right nodule 2 |
— |
Het |
— |
— |
— |
Het |
Homo |
— |
Het |
— |
— |
— |
— |
— |
| *II11 |
— |
Homo |
— |
— |
— |
— |
Het |
— |
Homo |
— |
— |
Het |
— |
— |
| *II11 right nodules |
— |
Homo |
Het |
— |
— |
Het |
Het |
— |
Homo |
— |
— |
— |
— |
— |
| *II13 |
— |
Het |
— |
— |
— |
— |
Het |
— |
Het |
— |
— |
— |
— |
— |
| *II15 |
— |
Homo |
— |
— |
— |
— |
Het |
— |
Homo |
— |
— |
— |
— |
— |
| III5 |
— |
Homo |
— |
— |
Het |
— |
— |
— |
Het |
— |
— |
— |
— |
— |
| III15 |
— |
Het |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
— |
— |
— |
| III18 |
— |
Homo |
— |
— |
— |
— |
Het |
— |
Homo |
— |
— |
— |
— |
— |
| III19 |
— |
Homo |
— |
— |
— |
Het |
Het |
— |
Homo |
— |
— |
Het |
— |
— |
| III20 |
— |
Het |
— |
— |
Het |
Het |
— |
— |
— |
— |
— |
Het |
— |
— |
| III21 |
— |
Homo |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
— |
— |
— |
| Family II |
*II3 |
— |
— |
— |
— |
— |
— |
— |
Het |
— |
Het |
— |
— |
— |
— |
| *II3 nodules |
— |
— |
— |
Het |
— |
— |
— |
Het |
— |
Het |
— |
— |
— |
— |
| *III9 |
Het |
— |
— |
— |
— |
— |
— |
Het |
— |
Het |
— |
— |
— |
— |
| sporadic nodule tissues |
Nodule #1 |
— |
Homo |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
| Nodule #2 |
— |
Homo |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
Het |
| Nodule #3 |
— |
Homo |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
— |
— |
Het |
| Nodule #6 |
— |
Homo |
— |
— |
Homo |
— |
— |
— |
— |
— |
— |
— |
— |
Het |
* denotes diagnosed patient of the two families; – indicates homozygous wild-type; † denotes co-segregated nsSNV of ARMC5 in the first family; § denotes pre-mature stop codon of ARMC5 of the second family.
Discussion
Fig. 6 showed an integrated ARMC5 mutation spectrum. It is noteworthy that except for the germ-line R315W mutation in exon 3, the right nodule 1 of patient II9 from the first family didn’t contain the 22-nt exon 1 deletion as that in patient II11’s nodule. Instead, a heterozygous A224E mutation was observed on the distinct haplotype. Variant effect prediction by SIFT and PolyPhen-2 suggested that this substitution is damaging. This fact had two implications: firstly, the second-hit mutation of different adrenal cell might be distinct and independent. Secondly, individuals with the A224E mutation might develop adrenal hyperplasia phenotype later on. That is to say, follow-up visit of the first family member with either R315W mutation or A224E mutation (III18, III19, and III20) should be necessary. Specifically, blood DNA of III4 had both of these variants, which calls for further attention. Moreover, homozygosity of R315W in the right nodule 2 of patient II9 in family I, on the other hand, seemed to result from LOH rather than de novo somatic mutation. Despite the reported wide occurrence of somatic LOH in AIMAH patients [9], four adrenal nodules involved in this study had no signature of LOH along the ARMC5 gene.
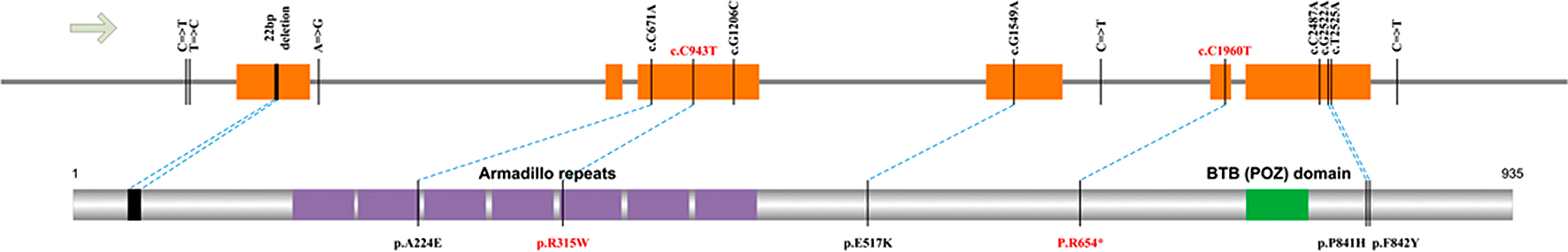
Of this study, novel germline variants in ARMC5 gene with familial AIMAH have been identified using whole genome sequencing (WGA), one of which had been reported in other research about AIMAH [23]. Biochemical and secretagogue examination of AIMAH could not provide the early diagnosis of one germline ARMC5 mutation. Previous studies mainly consider AIMAH as one incidental disease. However, a series of evidences suggest that hereditary factor play important role in this disease [23, 24]. Firstly, the mutations of ARMC5 gene could lead to the potential tumorigenesis during embryogenesis. Secondly, the results of carrier screening of normal phenotype individuals, which are relatives of patients with AIMAH, indicated that they are disease-causing mutation carriers [25]. Based on the results mentioned above, we speculated that mutations of AIMAH gene could be more widespread in the whole pedigree than the prior reorganization. Thirdly, long term of tumorigenesis, hypercortisolism and the late ageat would prevent the recognized of this genetics disease [26]. Last but not the least, two recently studies all have suggested that about half of in-group members owned mutations in ARMC5 gene, which is the powerful evidence for the AIMAH characters of family genetic [7].
We also notice an interesting phenomenon that mutations of ARMC5 alone seem to be insufficient to lead to AIMAH. Therefore, second-hit mutations model have been provided to explain the patients with genetic mutations of ARMC5 gene [23]. The second-hit mutations have been found in different tumor nodules originated from one adrenal gland, which could reflect the heterogeneity of AIMAH [7]. On the other hand, the slow progression of adrenal nodularity, the endogenic onset of hypercortisolism, and the late age could all be understood by environmental factors and the second-hit mutation model [27].
The ARMC5 gene mutations in AIMAH patients would provide an important roadmap for the clinical care of AIMAH families. For example, mutations in ARMC5 gene could be screened out in abnormal phenotype individuals, and these mutations would be helpful to carrier screening and clinical diagnostic. Therefore, the most urgent thing would be draw the mutation spectrum through larger-scale screening of AIMAH patients. Furthermore, the mechanism of ARMC5 mutations related tumorigenesis and somatic second-hit mutation are still confused. Therefore, It is worthy to testing the status of ARMC5 mutation of patients with adrenal gland related cancer. Meanwhile, there still is poor knowledge about the accurate penetrance of germline ARMC5 mutations. In future, long-term studies of various ARMC5 mutations should be performed to decrypt the related information. The last but not the least, the molecular mechanism of AIMAH in ARMC5 mutation-negative patients would also need a lot of works [12, 18].
This study broadens the atlas of ARMC5 mutations with potential familial AIMAH. WGA method would be an effective mean to screen and identify candidate genes and mutations. Others have indicated there is none of the absolute correspondence relationships between not AIMAH and ARMC5 mutations [18]. However, affected members of this study will provide the powerful evidences because the genetic mutation is identical.
Abbreviations
AIMAH, ACTH-independent macronodular adrenal hyperplasia; WGA, whole-genome sequencing; CS, Cushing’s syndrome; ARMC5, armadillo repeat containing 5; CT, computed tomography; SCS, sub-clinical features of CS
Authors’ Contributions
Q.Z. and L.C. for bioinformatics analysis and writing of the manuscript. J.P.G., L.Z. and J.M.L. for discussion and comments on an earlier version of the manuscript. All authors read and approved the final manuscript.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1 Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, et al. (2013) Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 369: 2115–2125.
- 2 Bourdeau I, Boisselle A, Rioux D, et al. (2007) Systematic clinical screening of members of a family with hereditary cortisol-secreting B-adrenergic responsive ACTH-independent macronodular adrenal hyperplasia (AIMAH) reveals unsuspected subclinical Cushing’s syndrome (CS) and aberrant B-adrenergic regulation of cortisol secretion. Program of the 89th Annual Meeting of the Endocrine Society. Toronto, CA p. 148, OR54-2 (Abstract).
- 3 Lacroix A, Ndiaye N, Tremblay J, Hamet P (2001) Ectopic and abnormal hormone receptors in adrenal Cushing’s syndrome. Endocr Rev 22: 75–110.
- 4 Mircescu H, Jilwan J, N’Diaye N, Bourdeau I, Tremblay J, et al. (2000) Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing’s syndrome? J Clin Endocrinol Metab 85: 3531–3536.
- 5 Hsiao HP, Verma S, Boikos SA, et al. (2007) A molecular and clinical genetic investigation of ACTH-independent macronodular adrenal hyperplasia compared to other, common adrenocortical tumors: evidence for heterogeneity, overlap with other tumor syndromes and frequent but atypical hormonal secretion. Program of the 89th Annual Meeting of the Endocrine Society. Toronto, p. 403, P2–299 (Abstract).
- 6 Aiba M, Kawakami M, Ito Y, Fujimoto Y, Suda T, et al. (1992) Bilateral adrenocortical adenomas causing Cushing’s syndrome. Report of two cases with enzyme histochemical and ultrastructural studies and a review of the literature. Arch Pathol Lab Med 116: 146–150.
- 7 Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, et al. (2013) ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med 369: 2105–2114.
- 8 Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, et al. (2014) Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab 99: E1113–E1119.
- 9 Aiba M, Hirayama A, Iri H, Ito Y , Fujimoto Y, et al. (1991) Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia as a distinct subtype of Cushing’s syndrome. Enzyme histochemical and ultrastructural study of four cases with a review of the literature. Am J Clin Pathol 96: 334–340.
- 10 Tewari R, Bailes E, Bunting KA, Coates JC (2010) Armadillo-repeat protein functions: questions for little creatures. Trends Cell Biol 20: 470–481.
- 11 Berthon A, Stratakis CA (2014) From b-catenine to ARM-repeat proteins in adrenocortical disorders. Horm Metab Res 46: 889–896.
- 12 Alencar GA, Lerario AM, Nishi MY, Mariani BM, Almeida MQ, et al. (2014) ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab 99: E1501–E1509.
- 13 Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, et al. (1991) Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325: 1688–1695.
- 14 Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, et al. (2000) Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 26: 89–92.
- 15 Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, et al. (2006) A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 38: 794–800.
- 16 Horvath A, Mericq V, Stratakis CA (2008) Mutation in PDE8B, a cAMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med 358: 750–752.
- 17 Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, et al. (1997) Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276: 404–407.
- 18 Cao Y, He M, Gao Z, Peng Y, Li Y, et al. (2014) Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 344: 913–917.
- 19 Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, et al. (2014) Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344: 917–920.
- 20 Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, et al. (2014) Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet 46: 613–617.
- 21 Katoh M, Katoh M (2004) Human FOX gene family. Int J Oncol 25: 1495–1500.
- 22 Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68: 820–823.
- 23 Lee S, Hwang R, Lee J, Rhee Y, Kim DJ, et al. (2005) Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin Endocrinol (Oxf) 63: 625–630.
- 24 Nies C, Bartsch DK, Ehlenz K, Wild A, Langer P, et al. (2002) Familial ACTH-independent Cushing’s syndrome with bilateral macronodular adrenal hyperplasia clinically affecting only female family members. Exp Clin Endocrinol Diabetes 110: 277–283.
- 25 Gagliardi L, Hotu C, Casey G, Braund WJ, Ling KH, et al. (2009) Familial vasopressin-sensitive ACTH-independent macronodular adrenal hyperplasia (VPs-AIMAH): clinical studies of three kindreds. Clin Endocrinol (Oxf) 70: 883–891.
- 26 N’Diaye N, Hamet P, Tremblay J, Boutin JM, Gaboury L, et al. (1999) Asynchronous development of bilateral nodular adrenal hyperplasia in gastric inhibitory polypeptide-dependent Cushing’s syndrome. J Clin Endocrinol Metab 84: 2616–2622.
- 27 Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, et al. (2009) Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab 94: 2930–2937.

