Abstract
Monocarboxylate transporter 8 (MCT8) facilitates T3 uptake into cells. Mutations in MCT8 lead to Allan-Herndon-Dudley syndrome (AHDS), which is characterized by severe psychomotor retardation and abnormal thyroid hormone profile. Nine uncharacterized MCT8 mutations in Japanese patients with severe neurocognitive impairment and elevated serum T3 levels were studied regarding the transport of T3. Human MCT8 (hMCT8) function was studied in wild-type (WT) or mutant hMCT8-transfected human placental choriocarcinoma cells (JEG3) by visualizing the locations of the proteins in the cells, detecting specific proteins, and measuring T3 uptake. We identified 6 missense (p.Arg445Ser, p.Asp498Asn, p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, and p.Gly312Arg), 2 frameshift (p.Arg355Profs*64 and p.Tyr550Serfs*17), and 1 deletion (p.Pro561del) mutation(s) in the hMCT8 gene. All patients exhibited clinical characteristics of AHDS with high free T3, low-normal free T4, and normal-elevated TSH levels. All tested mutants were expressed at the protein level, except p.Arg355Profs*64 and p.Tyr550Serfs*17, which were truncated, and were inactive in T3 uptake, excluding p.Arg445Ser and p.Pro561del mutants, compared with WT-hMCT8. Immunocytochemistry revealed plasma membrane localization of p.Arg445Ser and p.Pro561del mutants similar with WT-hMCT8. The other mutants failed to localize in significant amount(s) in the plasma membrane and instead localized in the cytoplasm. These data indicate that p.Arg445Ser and p.Pro561del mutants preserve residual function, whereas p.Asp498Asn, p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, p.Gly312Arg, p.Arg355Profs*64, and p.Tyr550Serfs*17 mutants lack function. These findings suggest that the mutations in MCT8 cause loss of function by reducing protein expression, impairing trafficking of protein to plasma membrane, and disrupting substrate channel.
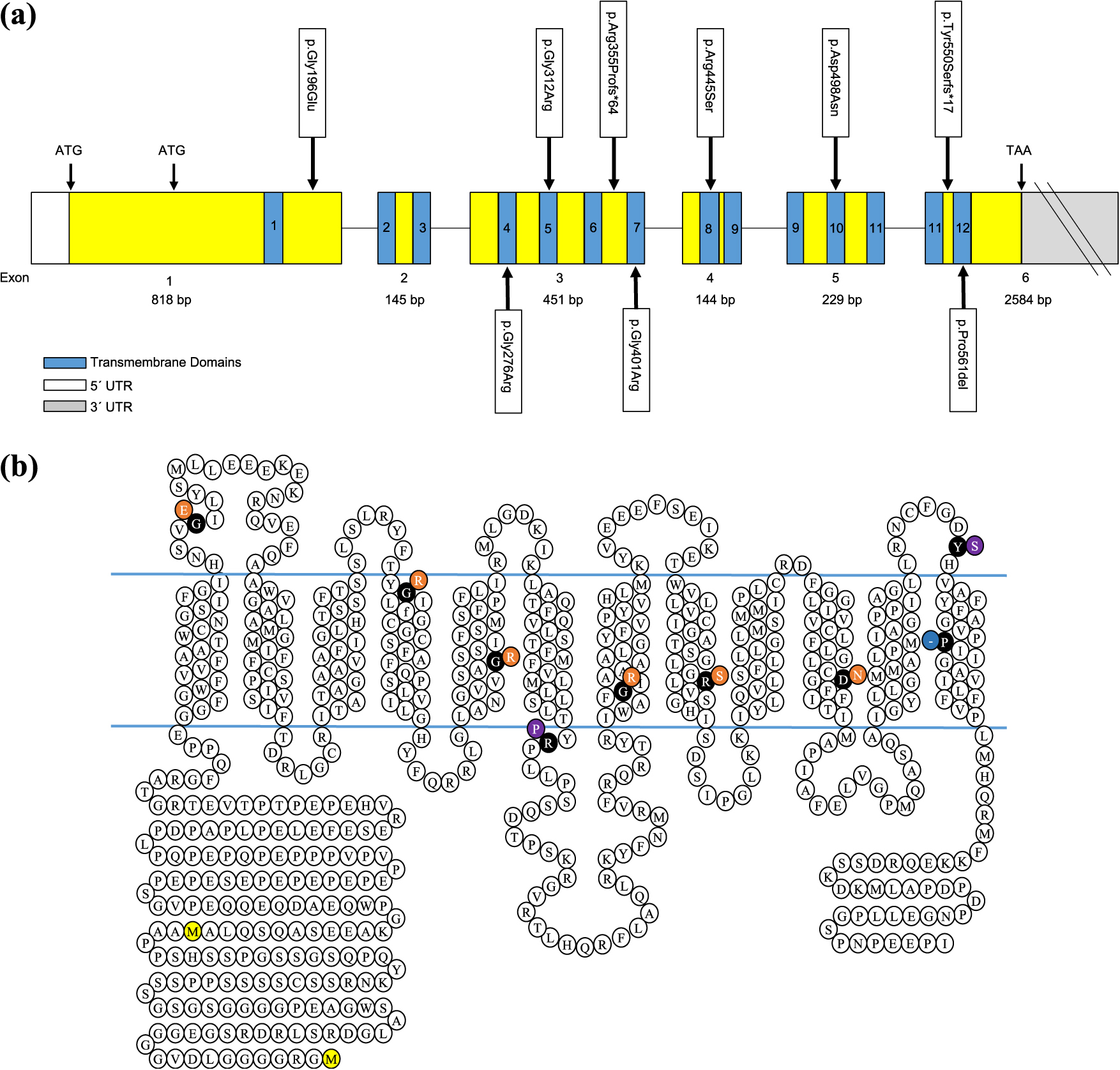
THYROID HORMONE (TH) plays a vital role in the normal growth and development of different tissues, such as bone, and especially in the human brain [1-4]. Transport of T3 across the cell membrane is essential because the metabolism and activity of TH occur intracellularly [5]. Several TH transporters, organic anion transporting polypeptide (OATP1C1), monocarboxylate transporter 8 (MCT8), and monocarboxylate transporter 10 (MCT10) are involved in this process. MCT8 (MIM# 300095) is a highly specific transporter that transports TH across the blood-brain barrier and into neuronal cells [6-13]. MCT8, also referred to as solute carrier family 16 member 2 (SLC16A2), is located on the human X-chromosome (Xq13.2) and consists of twelve transmembrane domains (TMDs) with six exons. It encodes two potential proteins of 539 and 613 amino acids depending on the use of two alternative translational start sites in exon 1 [13-15]. The short MCT8 protein is thought to be physiologically functional [16]. MCT8 is highly expressed in the brain and liver, and is expressed in some other tissues, including the kidneys and thyroid [17-20].
Hemizygous SLC16A2 loss-of-function mutations in males lead to a rare X-linked disorder called Allan-Herndon-Dudley syndrome (AHDS) (OMIM #300523), which is characterized by severe psychomotor retardation and an abnormal TH profile with high serum T3, low-normal T4, low reverse T3 (rT3), and normal-elevated TSH levels [1, 21, 22]. Psychomotor retardation encompasses several pathological conditions such as moderate-severe cognitive impairment, difficulties or inabilities in sitting, walking or standing alone, dysarthria-completely absent speech, infantile hypotonia evolving to spastic paraplegia, and gradual development of spasticity [1, 3, 13, 23-26]. Although the pathogenic mechanism of MCT8 mutation remains unclear, psychomotor retardation is considered to be due to the lack of TH transporter function, which leads to a hypothyroid state in the brain and subsequent brain damage [27, 28]. The changes in TH secretion and an increased peripheral conversion of T4 to T3 cause the abnormal TH profile [29-31].
To date, over 100 mutations in the SLC16A2 gene have been reported in patients with AHDS, including missense, nonsense, deletion, insertion, frame shift, and splice site mutations [1, 32]. The pathogenic mechanism varies even though most of the mutations cause complete inactivation of TH influx/efflux activity [21, 22]. Most clinically relevant mutations, such as truncating mutations, insertions/deletions, and missense mutations, interfere with proline or glycine residues, which are predicted to be located within TMDs and affect protein expression as well as subcellular localization. On the other hand, some mutations interfere with substrate translocation, whereas others affect protein trafficking and stability [6, 15, 33-36]. Detailed insight into the MCT8 structure is important to clarify the physiological substrate translocation mechanism and the impact of different mutations because protein structure and functions are intimately related. There are no crystal or nuclear magnetic resonance structures of MCT8 available, but protein homology modeling is an effective alternative [37].
In this study, we evaluated the function of 9 uncharacterized MCT8 mutations, 5 of which are novel, and described psychomotor findings as well as the TH profiles of the AHDS patients.
Materials and Methods
Serum and DNA analyses
The ‘Research Ethics Committee’ of Osaka University approved (#602) these studies and informed consent was obtained from the parents of each patient. Serum free T3 (fT3), free T4 (fT4), and TSH levels were measured at the patients’ respective hospital laboratories. The sequencing analysis of the MCT8 gene was performed using intronic primers flanking the 6 exons.
Plasmids
Wild–type human MCT8 (WT-hMCT8) was cloned into the expression vector pcDNA3.1 (empty vector), as described previously [28]. The mutations identified in patients were generated using In-Fusion HD Cloning Protocol (Takara Bio USA, Inc., USA) to produce the mutant proteins p.Arg445Ser, p.Pro561del, p.Asp498Asn, p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, p.Gly312Arg, p.Arg355Profs*64, and p.Tyr550Serfs*17. Since MCT8 mutations have traditionally been numbered based on the long MCT8 protein, we adhered to this system. Mutagenesis primers were purchased from Fasmac Co. Ltd., Japan. Chemically-competent Escherichia Coli were used for transformation followed by overnight incubation on Luria-Bertani (LB)-agar-plates containing 100 μg/mL ampicillin at 37°C. After selecting single clones, incubation was performed in selective S.O.C. (super optimized broth with catabolite repression) medium overnight at 37°C. Extraction of expression vectors was accomplished using the QiaPrep® Spin Miniprep Kit (Qiagen, Germany). Generation of mutations and absence of unintended mutations were confirmed by Sanger sequencing.
Cell culture and transfection
Human placental choriocarcinoma cells (JEG3, derived from human placental choriocarcinoma) (ATCC, USA) were cultured at 37°C with 5% CO2 in D-MEM/Ham’s F-12 medium (Wako Pure Chemical Industries Ltd., Japan), containing 9% heat-inactivated fetal bovine serum (Invitrogen, USA) and 100 nM sodium selenite (Sigma-Aldrich, USA). Transfections were carried out at 30–40% confluency, and all experiments were performed 48 h after transfection. Cells were seeded and transfected in duplicate or triplicate in 6-well dishes with 500 ng of WT, mutant-hMCT8s, or pcDNA3.1 for western blotting (WB) and the T3 uptake assay. For the immunocytochemistry assay, 200 ng of WT, mutant-hMCT8s, or empty vector were used for transfection. All transfections were carried out with a 1:3 ratio of plasmid DNA and Fu-Gene-6 transfection reagent (Roche Applied Science, The Netherlands) according to the manufacturer’s protocol. There were no significant differences in transfection efficiency among the MCT8 mutants as confirmed by co-transfection with green fluorescent protein (GFP) (data not shown).
T3 uptake
JEG3 cells were cultured in 6-well culture dishes and transfected in triplicate with 500 ng empty vector, WT, or mutant-hMCT8s. Forty-eight hours after transfection, cells were washed with 2 mL of D-MEM/Ham’s F-12 medium plus 0.1% BSA. Then, samples were incubated for 15 min at 37°C in 5% CO2 with 1 nM (≈1 × 106 cpm) [125I]T3 (PerkinElmer, Japan) in 1 mL of incubation medium. Cells were lysed with 1 mL of 0.1 M NaOH to count in a γ-counter (Packard Cobra II Gamma Counter, Model 5003, USA) after washing with incubation medium as described previously [35]. Statistical analysis was performed after deducting the background value and considering [125I]T3 uptake by WT-hMCT8 as 100%.
Western blotting
Transiently transfected (500 ng of WT, mutant-hMCT8s, or pcDNA3.1) JEG3 cells in 6-well culture dishes were harvested 48 h after transfection and washed with D-PBS. Sonication was performed after adding protease inhibitor and phosphatase inhibitor cocktail (Nacalai Tesque Inc., Japan) with lysis buffer. Then, 20 μg of the lysates was mixed with an appropriate amount of 2 × Lameli sample buffer and 2-mercaptoethanol (Bio-Rad Laboratories, Inc., USA), and denatured at 95°C for 5 min. Samples were next separated on a 7.5% SDS-PAGE gel and transferred to PVDF membranes (Bio-Rad Laboratories, Inc., USA). Finally, samples were probed with rabbit anti-hSLC16A2 antibody (Sigma-Aldrich, USA, code # HPA003353, 1:1,000) as described previously [38]. The house keeping protein Anti-β-Actin pAb-HRP-DirecT (MBL CO., LTD, Japan, 1:2,000) was used as a loading control and the expected band size for WT-hMCT8 was ~60 KDa.
The intensity of bands on WB of transiently transfected JEG3 cells with WT, mutant-hMCT8s, or pcDNA3.1 was measured by ImageJ software with hMCT8/β-actin = {(hMCT8/β-actin) – (pcDNA3.1/β-actin)}/(WT-hMCT8/β-actin). The background intensity was subtracted from the intensity of WT-hMCT8 and each of the mutants before graphical presentation. Statistical analysis was performed considering the intensity of WT-hMCT8 to be ‘1’.
Immunocytochemistry
JEG3 cells were cultured on 13-mm glass coverslips coated with poly-L-lysine (Matsunami Glass Industry Co. Ltd., Japan) in 12-well plates with 200 ng of pcDNA3.1, WT, or mutant-hMCT8s. Cells were fixed with 4% paraformaldehyde 48 h after transfection and permeabilized with 0.2% Triton X-100 in PBS. After blocking for 30 min with PBS containing 2% BSA, samples were stained with rabbit anti-hMCT8 antibody 1306 (a kind gift from Prof. Theo J. Visser, Erasmus University Medical Center, 1:1,000) and monoclonal mouse anti-zona occludens protein 1 (ZO-1; tight junction protein 1) antibody conjugated with Alexa Fluor 555 (Red) (Invitrogen, USA, 1:250). Coverslips were mounted with Prolong Gold antifade reagent with 4,6-diamidino-2-phenylindole (DAPI, Blue) (Invitrogen, USA) after secondary staining with donkey antirabbit Alexa Fluor 488 (Green) (Jackson ImmunoResearch laboratories, Inc., USA). Samples were examined on a Zeiss confocal laser scanning microscope LSM 710 using Zeiss LSM software ZEN 2.3 lite (Carl Zeiss Microscopy GmbH, Germany) as described previously [38].
We also calculated the percentage of cells expressing hMCT8 protein in the plasma membrane. The percentage was calculated by considering the total number of cells expressing hMCT8 protein.
3D structure model
3D WT or mutant-hMCT8 structures were generated using SWISS-MODEL from the https://swissmodel.expasy.org/interactive.
Statistical analysis
The results of all experiments are presented as means and standard error of means (SEM) of 3 separate experiments performed in duplicate or triplicate. Statistical analysis was carried out with JMP pro 13 software for Windows (SAS, USA). The significance of differences among WT-hMCT8, mutant-hMCT8 and pcDNA3.1 was tested using one-way ANOVA followed by the Tukey HSD test. P < 0.05 was considered significant.
Results
We identified 9 uncharacterized MCT8 gene mutations in 13 boys of differing ages (6M to 20Y) with severe mental retardation. Three mutations were found in more than one patient. Although most patients had hypotonia and dyskinetic cerebral palsy, seizures were less common. The TSH level was normal to elevated [Normal range (NR): 0.4–3.8 μIU/mL], whereas serum fT3 was markedly elevated (NR: 2–3.4 pg/mL) and fT4 was below the normal range (NR: 0.9–1.6 ng/dL) (Table 1).
Table 1
Mutations, clinical phenotype and thyroid hormone serum levels of MCT8 patients at the first visit
| Mutation |
Protein |
Number of Patients |
Patient’s Age |
Mental Retardation |
Sit Unassisted/Pull up |
Hypotonia |
Dyskinetic Cerebral Palsy |
Seizure |
TSH (μIU/mL) |
fT3 (pg/mL) |
fT4 (ng/dL) |
| c.1333C>A |
p.Arg445Ser |
1 |
8Y |
+ |
N/A |
+ |
N/A |
– |
2.44 |
4.20 |
0.60 |
| c.1681_1683delCCC |
p.Pro561del |
2 |
(a) 7Y |
+ |
+ |
N/A |
+ |
+ |
3.00 |
5.56 |
0.607 |
| (b) 11M |
+ |
N/A |
+ |
N/A |
– |
7.67 |
7.26 |
0.81 |
| c.1492G>A |
p.Asp498Asn |
1 |
11M |
+ |
N/A |
+ |
+ |
– |
5.95 |
6.78 |
0.52 |
| c.826G>A |
p.Gly276Arg |
3 |
(a) 6M |
+ |
– |
+ |
+ |
– |
4.09 |
7.50 |
0.70 |
| (b) 1Y |
+ |
– |
+ |
+ |
– |
3.29 |
9.22 |
0.64 |
| (c) 1Y |
+ |
– |
+ |
+ |
– |
1.55 |
6.60 |
0.65 |
| c.587G>A |
p.Gly196Glu |
1 |
19Y |
+ |
N/A |
+ |
N/A |
– |
7.09 |
6.20 |
0.30 |
| c.1201G>A |
p.Gly401Arg |
1 |
2Y |
+ |
– |
+ |
+ |
+ |
1.73 |
7.80 |
0.50 |
| c.934G>A |
p.Gly312Arg |
2 |
(a) 1Y |
+ |
N/A |
N/A |
+ |
– |
5.05 |
8.53 |
0.65 |
| (b) 1Y |
+ |
N/A |
N/A |
+ |
– |
3.97 |
9.67 |
0.66 |
| c.1063_1064insCTACC |
p.Arg355Profs*64 |
1 |
20Y |
+ |
N/A |
+ |
N/A |
+ |
1.26 |
5.60 |
0.70 |
| c.1649delA |
p.Tyr550Serfs*17 |
1 |
3Y |
+ |
– |
+ |
+ |
– |
4.00 |
5.50 |
0.50 |
+, Present; –, Absent; N/A, Not available
All 9 MCT8 mutations, 5 of which were novel, were different hemizygous mutations. p.Arg445Ser, p.Asp498Asn, p.Gly276Arg, p.Gly401Arg, and p.Gly312Arg were missense mutations and located in TMDs-8, 10, 4, 7, and 5 of exons 4, 5, 3, 3, and 3, respectively. p.Gly196Glu was also a missense mutation in the topological domain of exon 1. TMD-12 of exon 6 contained p.Pro561del, a deletion mutation. Two frameshift mutations, p.Arg355Profs*64 and p.Tyr550Serfs*17, were located in the topological domain of exon 3 and 6, respectively (Fig. 1a and 1b).
Uptake of T3
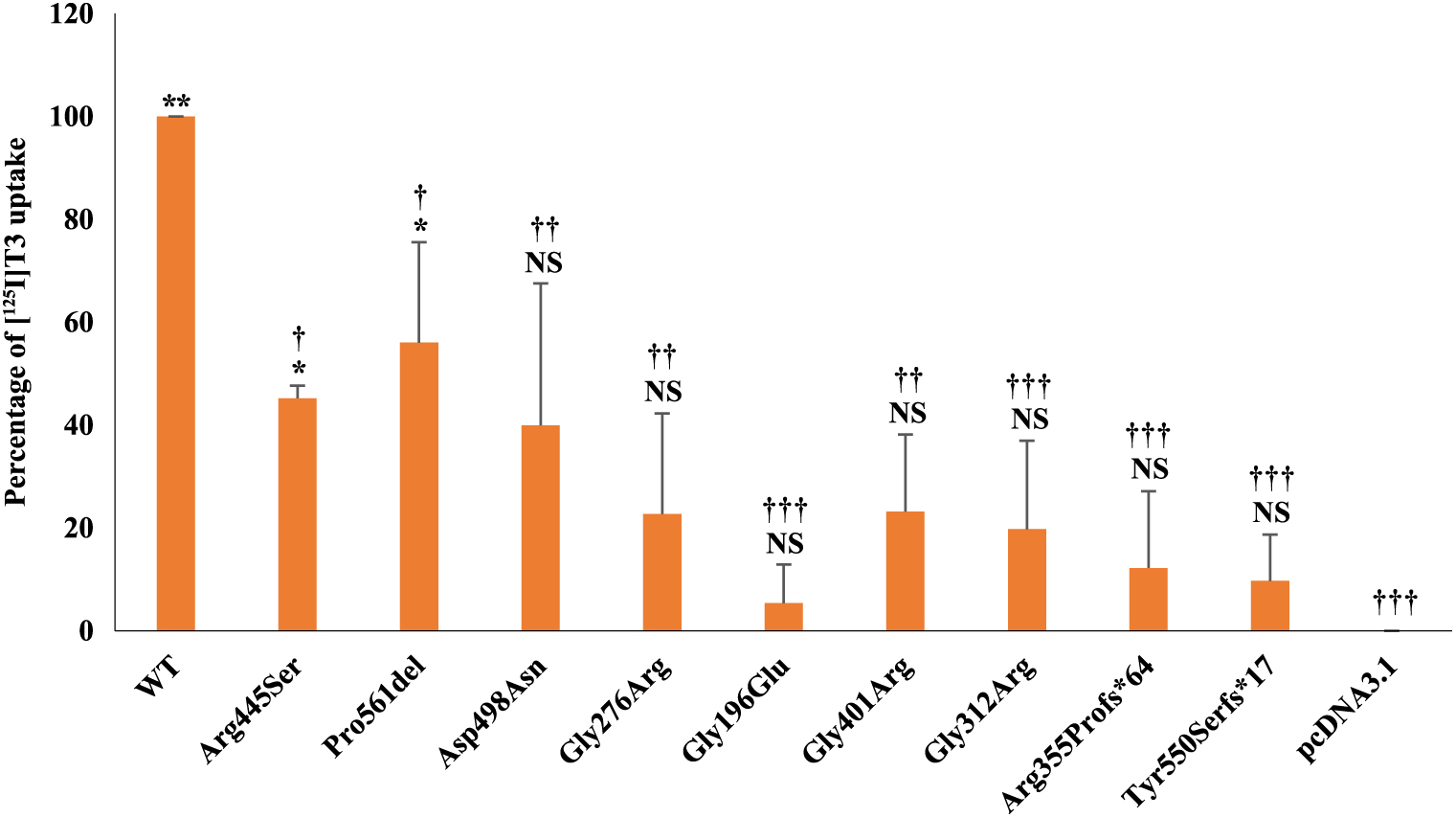
We analyzed the 15-minute uptake of [125I]T3 after transiently transfecting JEG3 cells with WT, mutant-hMCT8s, or pcDNA3.1 and incubating for 48 h at 37°C (Fig. 2). Although JEG3 cells do not express endogenous MCT8 [19], pcDNA3.1-transfected cells exhibited significant [125I]T3 uptake. This background was likely due to other transporter(s). Transfection of cells with p.Arg445Ser and p.Pro561del resulted in statistically significant increased [125I]T3 uptake, 45.21 and 56.04%, respectively, compared with pcDNA3.1. p.Asp498Asn had almost the same activity (39.93%) as p.Arg445Ser, but was not statistically significant different from that of the empty vector. The uptake of the other mutants p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, p.Gly312Arg, p.Arg355Profs*64, and p.Tyr550Serfs*17 were not significantly different from pcDNA3.1.
Western blotting
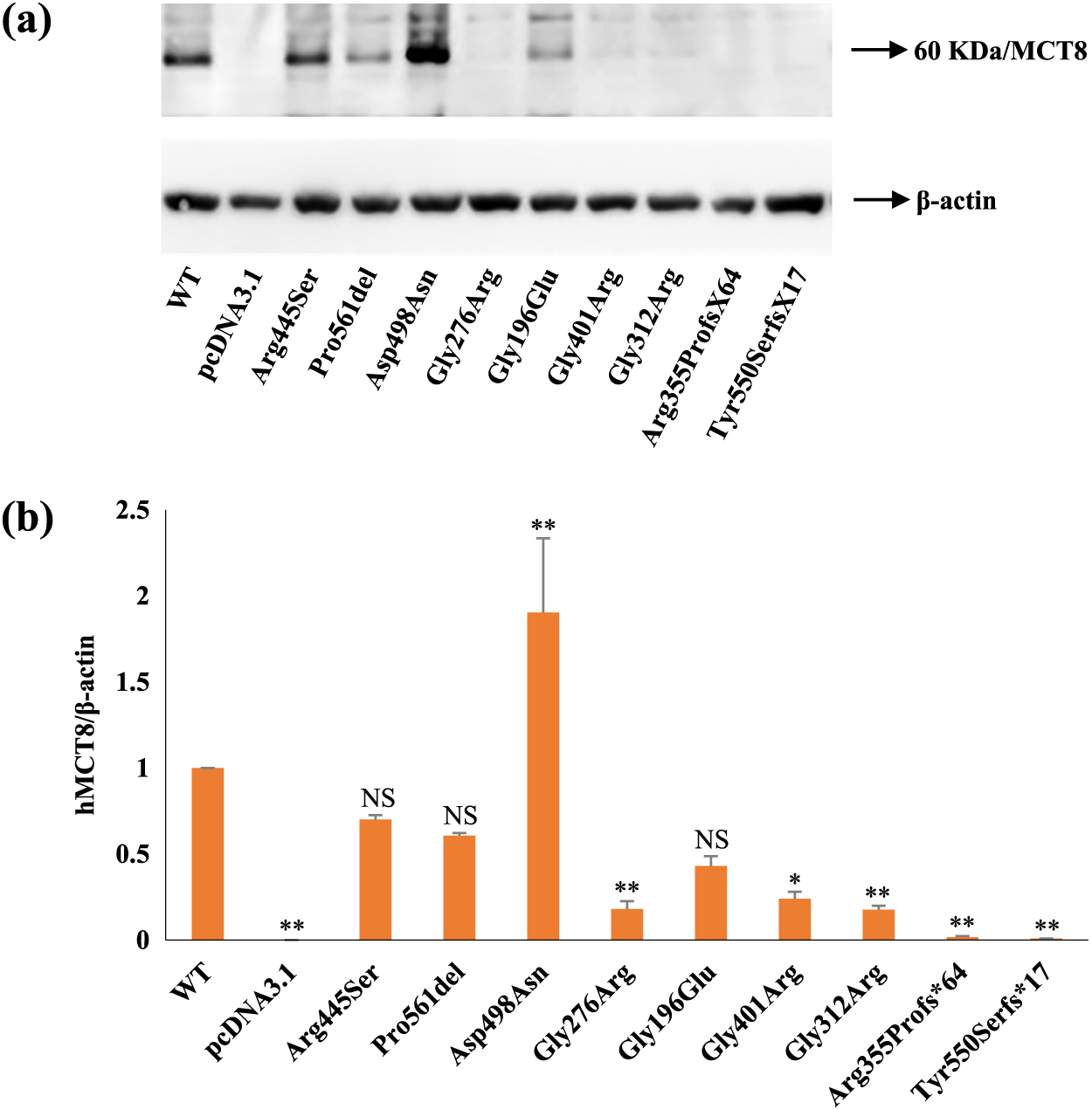
Immunoblotting of whole-cell lysates from transiently transfected JEG3 cells with WT, mutant-hMCT8s, or pcDNA3.1 is shown in Fig. 3a. p.Arg445Ser was expressed at similar levels, whereas p.Asp498Asn was expressed at much higher levels but at the same size as WT-hMCT8. Expression levels of p.Pro561del and p.Gly196Glu were similar but slightly lower than that of WT-hMCT8. Low expression was detected for p.Gly401Arg- and p.Gly312Arg-transfected cells and a much weaker band was observed for p.Gly276Arg compared with WT-hMCT8. The expected bands of the two frameshift mutations p.Arg355Profs*64 and p.Tyr550Serfs*17 were not detected on WB. JEG3 cells transfected with pcDNA3.1 also failed to produce a band at the expected size. Furthermore, we measured the intensity of bands from the WB assay performed with JEG3 cells transiently transfected with WT, mutant-hMCT8s, or pcDNA3.1 (Fig. 3b). p.Arg445Ser had a band intensity with a value of 0.7 compared with that of WT-hMCT8. p.Pro561del and p.Gly196Glu produced a moderate to low band intensity of 0.6 and 0.43, respectively, in comparison with WT-hMCT8. p.Asp498Asn had a significant band intensity of 1.90, which was greater than the WT-hMCT8. p.Gly276Arg, p.Gly401Arg, and p.Gly312Arg showed negligible band intensities compared to WT-hMCT8. p.Arg355Profs*64 and p.Tyr550Serfs*17 had immeasurable band intensities.
We next evaluated the localization of mutant-hMCT8s by immunocytochemistry (Fig. 4a). No specific hMCT8 staining was observed in cells transfected with pcDNA3.1. Clear co-localization of WT-hMCT8 with the plasma membrane marker ZO-1 indicated expression at the plasma membrane. p.Arg445Ser and p.Pro561del exhibited partial co-localization with the plasma membrane marker, suggesting expression of these mutants at the plasma membrane. In contrast, minimal co-localization with the plasma membrane marker was noted for p.Asp498Asn, p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, and p.Gly312Arg, indicating a defect in trafficking of these mutants to the plasma membrane. hMCT8-specific staining for p.Arg355Profs*64 and p.Tyr550Serfs*17 was not detectable by our antibody. We also calculated the percentage of cells expressing WT or mutant-hMCT8 proteins on the plasma membrane (Fig. 4b). While comparison of mutants-hMCT8s with WT-hMCT8 shows that all the differences are statistically significant, we observed a higher percentage of cells expressing MCT8 on the plasma membrane in p.Arg445Ser and p.Pro561del. A non-significant difference between p.Arg445Ser and p.Pro561del, and a significant difference between p.Arg445Ser/p.Pro561del and all the other mutants were noted in percentage of cells expressing MCT8 on the plasma membrane.

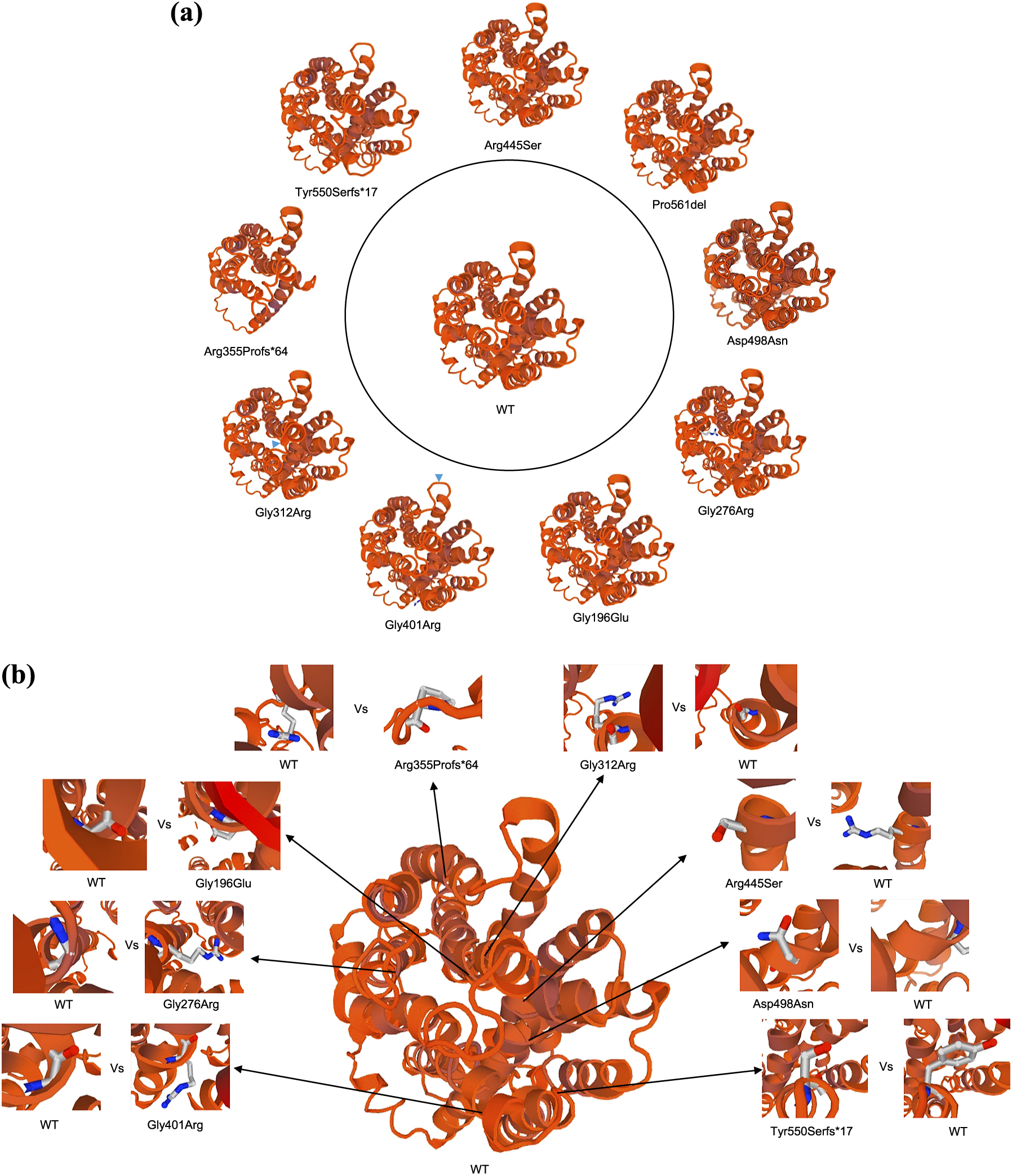
We constructed 3D WT and mutant-hMCT8 protein structures (Fig. 5a). p.Gly401Arg and p.Gly312Arg had shown alterations of the helices (Fig. 5a). Moreover, p.Arg355Profs*64 and p.Tyr550Serfs*17 had clear structural differences from WT-hMCT8. On the other hand, p.Arg445Ser, p.Pro561del, p.Asp498Asn, p.Gly276Arg, and p.Gly196Glu were not structurally different from WT-hMCT8.
Discussion
In the present study, we reported 13 patients with 9 uncharacterized mutations, 5 of which are novel in the MCT8 gene located on the X-chromosome. All patients had severe mental retardation with elevated serum T3 levels. We found that these mutations were either partially active or almost completely inactive based on the reduced uptake of T3 in vitro. Different mechanisms, such as impaired protein expression, defective trafficking to the plasma membrane, and presumably loss of substrate-MCT8 interaction were involved in this loss of function [4, 18, 35].
All mutant-hMCT8 proteins tested in this study were expressed in JEG3 cells at varying levels, except p.Arg355Profs*64 and p.Tyr550Serfs*17 because of truncation, demonstrating that impaired synthesis was not a major mechanism for the loss of function of the MCT8.
p.Pro561del, p.Gly276Arg, p.Gly196Glu, p.Gly401Arg, and p.Gly312Arg produced low levels of protein expression on WB compared with WT-hMCT8. Previous reports have shown that the difference in protein levels cannot be explained by impaired transcription. They pointed out that more rapid mutant protein degradation was responsible for the lower protein level, which leads to attenuated function [18, 38]. We assume that the same is also occurring with the mutants in our study. p.Asp498Asn showed elevated protein expression levels but exhibited minimal T3 uptake suggesting loss of the function of the protein itself.
Trafficking of the mature protein to the plasma membrane is very important for hMCT8 function [4, 18]. Immunocytochemistry revealed that although p.Arg445Ser and p.Pro561del mutant proteins have the ability to localize to the plasma membrane, the percentage of cells actually expressing the protein at the plasma membrane was less than that of WT-hMCT8. This indicates that the reduced T3 transport activity is at least in part due to less functional transporters at the plasma membrane, which is consistent with the reported partial activity of p.Arg271His [4]. All the other mutants including p.Asp498Asn and p.Gly196Glu with reasonable protein expression levels were minimally localized to the plasma membrane. These mutations most likely disturb protein folding, inhibit trafficking to the plasma membrane, and reduce the amount of protein expressed at the plasma membrane. Alternatively, minimal localization to the plasma membrane of p.Gly276Arg, p.Gly401Arg, p.Gly312Arg, p.Arg355Profs*64, and p.Tyr550Serfs*17, in particular, may be explained by very low or no expression of these mutant proteins.
According to a previous study, His192, Arg445, and Asp498 are presumed to constitute a substrate-binding center [37], which is crucial for TH transport. It is proposed that the interaction between TH and Arg445, and a His-Arg clamp formation between His192 and Arg445 are necessary for substrate recognition [33, 37]. Arg445 and Asp498 forms a hydrogen bond due to close structural proximity [6, 33, 34, 37]. Alterations in this interaction may lead to conformational changes by modifying the orientation of the Asp498 side chain/TMD-10 in the presence of TH [37]. A previous report has shown that natural or artificial substitutions of Arg445 render MCT8 functionally almost inactive [33]. This differs from our result that shows residual activity with a Ser mutation, indicating that transport properties may vary depending on the substituted amino acid. In the same paper, all tested mutations in Asp498 resulted in almost complete loss of function, which is consistent with our result. Depending on the relative orientation toward the substrate, it is assumed that other residues at/near the substrate-binding center have varying importance for substrate translocation [37].
A 3D model structure study has shown that pathogenic mutations can emerge from substitutions either at or to Pro/Gly [39]. In our 3D model, alterations of the helices in p.Gly312Arg and p.Gly401Arg were observed (Fig. 5a). In contrast, no apparent change was seen in p.Gly276Arg. All the three Glys are in alpha-helices. A close-up view shows that, in contrast to Gly whose side chain resides within the helix, substitution to Arg causes the side chain to protrude outward, which may disrupt association of the protein with the lipid bilayer (Fig. 5b). Intriguingly, p.Pro561del resides in a helix but shows considerable residual activity. We speculate that the protein structure is fairly well conserved since it is a deletion rather than a substitution.
One of the two patients harboring the p.Pro561del mutation was able to sit up by himself, supporting our observation of preserved partial MCT8 function, which is consistent with other reports [38, 40, 41]. The other patient (11M) was too young to assess these features. Although we could not confirm whether the patient with the p.Arg445Ser mutation can sit up, this protein also maintains residual activity. He was not able to walk at the age of 8 years [26]. The patients harboring the p.Gly276Arg and p.Gly401Arg mutations were unable to sit up and speak even 5–10 years after the initial diagnosis, which most probably points to severely limited MCT8 function. We have reported that the patient with the p.Tyr550Serfs*17 mutation was unable to sit or speak at the age of 2 years and 7 months [13]. He has not shown much development since then. The other patients with p.Gly196Glu and p.Arg355Profs*64 had clinical findings characteristics of AHDS [26] and revealed minimal MCT8 function. The patients harboring p.Asp498Asn (11M) and p.Gly312Arg (1Y) mutations were too young to assess phenotypic severity (Table 1).
In conclusion, our study underscores that the functional defects of mutant-hMCT8 proteins are the result of protein expression reduction, impairment in trafficking to the plasma membrane, substrate channel disruption, and modification of helices conformation. Mutant-hMCT8s that localize to the plasma membrane will more likely have residual activity and result in a less severe phenotype.
Note Added in Proof
While preparing the manuscript, we found that p.Asp498Asn had already been characterized by Groeneweg et al. [33].
Acknowledgements
We wish to thank all the primary doctors of the patients in our study. Dr. Miki Inutsuka, Department of Pediatrics, Sasebo Chuo Hospital; Dr. Satoshi Morita, Department of Pediatrics, Kawanabe Seikyo Hospital; Dr. Erina Ono, Department of Pediatrics, The Jikei University School of Medicine and Department of Pediatrics, Tokyo Metropolitan Kita Medical and Rehabilitation Center for the Disabled; Dr. Sumito Dateki, Department of Pediatrics, Nagasaki University Graduate School of Biomedical Sciences; Dr. Takako Fujita, Department of Pediatrics, Fukuoka University. The authors also wish to thank Prof. Theo J. Visser, Ph.D., Department of Internal Medicine, Erasmus University Medical Center, 3000 CA, Rotterdam, The Netherlands for his generous gift of rabbit anti-hMCT8 antibody 1306.
Disclosure
None of the authors have any potential conflicts of interest associated with this research.
References
- 1 Novara F, Groeneweg S, Freri E, Estienne M, Reho P, et al. (2017) Clinical and molecular characteristics of SLC16A2 (MCT8) mutations in three families with the Allan-Herndon-Dudley syndrome. Hum Mutat 38: 260–264.
- 2 Anik A, Kersseboom S, Demir K, Catli G, Yis U, et al. (2014) Psychomotor retardation caused by a defective thyroid hormone transporter: report of two families with different MCT8 mutations. Horm Res Paediatr 82: 261–271.
- 3 Visser WE, Vrijmoeth P, Visser FE, Arts WF, van Toor H, et al. (2013) Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin Endocrinol (Oxf) 78: 310–315.
- 4 Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, et al. (2007) Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab 92: 2378–2381.
- 5 Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, et al. (2001) Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 22: 451–476.
- 6 Kinne A, Kleinau G, Hoefig CS, Gruters A, Kohrle J, et al. (2010) Essential molecular determinants for thyroid hormone transport and first structural implications for monocarboxylate transporter 8. J Biol Chem 285: 28054–28063.
- 7 Heuer H, Visser TJ (2009) Minireview: pathophysiological importance of thyroid hormone transporters. Endocrinology 150: 1078–1083.
- 8 Wirth EK, Roth S, Blechschmidt C, Holter SM, Becker L, et al. (2009) Neuronal 3',3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci 29: 9439–9449.
- 9 Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, et al. (2009) Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3'-triiodo-l-thyronine. Endocrinology 150: 2491–2496.
- 10 Muller J, Heuer H (2014) Expression pattern of thyroid hormone transporters in the postnatal mouse brain. Front Endocrinol (Lausanne) 5: 92.
- 11 Allan W, Herndon C, Dudley FC (1944) Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic 48: 325–334.
- 12 Abe S, Namba N, Abe M, Fujiwara M, Aikawa T, et al. (2012) Monocarboxylate transporter 10 functions as a thyroid hormone transporter in chondrocytes. Endocrinology 153: 4049–4058.
- 13 Namba N, Etani Y, Kitaoka T, Nakamoto Y, Nakacho M, et al. (2008) Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr 167: 785–791.
- 14 Schwartz CE, Stevenson RE (2007) The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab 21: 307–321.
- 15 Friesema EC, Visser WE, Visser TJ (2010) Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol 322: 107–113.
- 16 Frints SGM, Lenzner S, Bauters M, Jensen LR, Van Esch H, et al. (2008) MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet 16: 1029–1037.
- 17 Friesema EC, Jansen J, Milici C, Visser TJ (2005) Thyroid hormone transporters. Vitam Horm 70: 137–167.
- 18 Kersseboom S, Kremers GJ, Friesema EC, Visser WE, Klootwijk W, et al. (2013) Mutations in MCT8 in patients with Allan-Herndon-Dudley-syndrome affecting its cellular distribution. Mol Endocrinol 27: 801–813.
- 19 Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, et al. (2006) Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab 2: 512–523.
- 20 Braun D, Schweizer U (2015) Efficient activation of pathogenic deltaPhe501 mutation in monocarboxylate transporter 8 by chemical and pharmacological chaperones. Endocrinology 156: 4720–4730.
- 21 Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S (2004) A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74: 168–175.
- 22 Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, et al. (2004) Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364: 1435–1437.
- 23 Bernal J, Guadano-Ferraz A, Morte B (2015) Thyroid hormone transporters-functions and clinical implications. Nat Rev Endocrinol 11: 406–417.
- 24 Boccone L, Dessi V, Meloni A, Loudianos G (2013) Allan-Herndon-Dudley syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. Eur J Med Genet 56: 207–210.
- 25 Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, et al. (2008) A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics 121: e199–e202.
- 26 Ono E, Ariga M, Oshima S, Hayakawa M, Imai M, et al. (2016) Three novel mutations of the MCT8 (SLC16A2) gene: individual and temporal variations of endocrinological and radiological features. Clin Pediatr Endocrinol 25: 23–35.
- 27 Bernal J (2007) Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab 3: 249–259.
- 28 Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH (2006) Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 20: 2761–2772.
- 29 Trajkovic-Arsic M, Muller J, Darras VM, Groba C, Lee S, et al. (2010) Impact of monocarboxylate transporter-8 deficiency on the hypothalamus-pituitary-thyroid axis in mice. Endocrinology 151: 5053–5062.
- 30 Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, et al. (2010) Consequences of monocarboxylate transporter 8 deficiency for renal transport and metabolism of thyroid hormones in mice. Endocrinology 151: 802–809.
- 31 Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, et al. (2010) Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest 120: 3377–3388.
- 32 Visser TJ (2016) Cellular Uptake of Thyroid Hormones. In: De Groot LJ (ed) Thyroid Disease- www.thyroidmanager.org, MDTEXT.COM,INC, South DARTMOUTH.
- 33 Groeneweg S, Friesema EC, Kersseboom S, Klootwijk W, Visser WE, et al. (2014) The role of Arg445 and Asp498 in the human thyroid hormone transporter MCT8. Endocrinology 155: 618–626.
- 34 Groeneweg S, Lima de Souza EC, Visser WE, Peeters RP, Visser TJ (2013) Importance of His192 in the human thyroid hormone transporter MCT8 for substrate recognition. Endocrinology 154: 2525–2532.
- 35 Kinne A, Roth S, Biebermann H, Kohrle J, Gruters A, et al. (2009) Surface translocation and tri-iodothyronine uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol 43: 263–271.
- 36 Armour CM, Kersseboom S, Yoon G, Visser TJ (2015) Further insights into the Allan-Herndon-Dudley syndrome: clinical and functional characterization of a novel MCT8 mutation. PLoS One 10: e0139343.
- 37 Groeneweg S, Lima de Souza EC, Meima ME, Peeters RP, Visser WE, et al. (2017) Outward-open model of thyroid hormone transporter monocarboxylate transporter 8 provides novel structural and functional insights. Endocrinology 158: 3292–3306.
- 38 Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ (2008) Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology 149: 2184–2190.
- 39 Kleinau G, Schweizer U, Kinne A, Kohrle J, Gruters A, et al. (2011) Insights into molecular properties of the human monocarboxylate transporter 8 by combining functional with structural information. Thyroid Res 4: S4.
- 40 Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, et al. (2005) Allan-Herndon-Dudley Syndrome and the Monocarboxylate Transporter 8 (MCT8) Gene. Am J Hum Genet 77: 41–53.
- 41 Capri Y, Friesema EC, Kersseboom S, Touraine R, Monnier A, et al. (2013) Relevance of different cellular models in determining the effects of mutations on SLC16A2/MCT8 thyroid hormone transporter function and genotype-phenotype correlation. Hum Mutat 34: 1018–1025.