REVIEW
The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases
2019 Volume 66 Issue 6 Pages 485-496

Details
2019 Volume 66 Issue 6 Pages 485-496
The tumor suppressor gene p53 is mutated in approximately more than 50% of human cancers. p53 is also referred to as the “cellular gatekeeper” or “guardian of the genome” because it protects the body from spreading mutated genome induced by various stress. When the cells receives stimuli such as DNA damage, oncogene activation, oxidative stress or undernutrition, p53 gives rise to a number of cellular responses, including cell cycle arrest, apoptosis, cellular senescence and metabolic adaptation. Related to energy metabolisms, it has been reported that p53 reduces glycolysis and enhances mitochondrial respiration. p53 is also involved in the regulation of other cellular metabolism and energy production systems: amino acid metabolism, fatty acid metabolism, nucleic acid metabolism, anti-oxidation, mitochondrial quality control, and autophagy. Moreover, recent studies have shown that p53 gene polymorphisms affect life expectancy and lifestyle-related disease such as type 2 diabetes, suggesting that there is a certain relationship between p53 function and metabolic disorders. In addition, mutant p53 protein does not only lose the tumor suppressor function, but it also gains novel oncogenic function and contributes to tumor development, involving cellular metabolism modification. Therefore, the importance of multifunctionality of p53, particularly with regard to intracellular metabolisms, arouses therapeutic interest and calls attention as the key molecule among cancer, lifestyle-related diseases and life expectancy.
Tumor suppressor gene p53 induces a variety of cellular responses, including cell cycle arrest, apoptosis, metabolic regulation, and cellular aging, depending on the degree of cellular stress. These responses are regulated through p53, a transcription factor that acts on downstream genes involved in many different physiological functions via post-translational modifications such as phosphorylation or acetylation (Fig. 1). As metabolomics analysis technology advances, including metabolome analysis and metabolic flux analysis, our understanding of the characteristics of tumor cell metabolism has improved. For example, energy metabolic dynamics in tumor cells differ greatly from those in normal cells: tumor cells generate energy through glycolysis, a metabolic characteristic known as the Warburg effect. p53 inhibits glycolysis and enhances oxidative phosphorylation (OXPHOS). It is also understood that metabolic dynamics in cancer cells differ drastically from normal cells in terms of fat and amino acid metabolism. Various studies have reported that p53 plays a primary role as a transcription factor in the control of intracellular metabolism by regulating the expression of metabolic genes and acting in concert with other metabolic regulatory systems. In this review, we outline the mechanisms of intracellular metabolic control by p53 in cancer cells and describe recent findings concerning the association of p53 single nucleotide polymorphisms (SNPs) with obesity and diabetes, as well as the relationship between mutant p53 “gain of function” mutations and metabolic changes.

p53 and cellular responses
The tumor suppressor gene p53 is activated by various types of cellular stress to cause numerous cellular responses, including cell cycle arrest, apoptosis, cellular aging, and intracellular metabolism regulation. As a transcription factor, p53 selectively activates downstream genes with varying functions depending on the type and level of cellular stress, which is thought to determine cell fate. UV, ultraviolet light; ROS, reactive oxygen species.
Cancer development is closely related to lifestyle characteristics, including eating habits. In recent years, p53 has been found to affect the onset of several lifestyle-related diseases, such as type 2 diabetes and obesity, via modulation of the regulation of metabolism at an individual person level [1, 2]. Correlative analyses of p53 gene polymorphisms have revealed statistical relationships not only with cancer but also with diabetes. The p53 codon 72 SNP (Arg 72 Pro) has previously been found to affect tumorigenesis and longevity [3, 4] and was also recently been correlated with the onset of type 2 diabetes [5, 6]. Arg-genotype is reported to more susceptible to apoptosis [7]. In a study using a murine model of Arg 72 Pro, obesity, non-alcoholic fatty liver disease (NAFLD) and diabetes were observed in Arg-genotype mice consuming a high-fat diet. Moreover, relationships between p53 downstream genes were observed among Cdkn1a, tumor necrosis factor (Tnf) and Niemann-Pick C1-Like 1 (Npc1l1), which is involved in cholesterol metabolism [8].
In normal cells, AMP-activated protein kinase (AMPK) acts as a sensor that detects the balance between the supply and demand of energy so that excessive energy is not generated continuously. The intracellular AMP/ATP ratio increases when cells are in energy starvation or increased energy consumption, which activates AMPK and enhances energy production [9]. p53 is activated by AMPK. Simultaneously, an increase in the AMP/ATP ratio decreases the effect of Akt-Mdm2, suppressing p53 degradation. Thus, activated p53, which detects changes in intracellular energy balance, adapts to the energy-starved state by regulating the energy production pathway and acts in numerous ways to produce a shift to a new homeostatic state.
Energy metabolism in tumor cells is markedly different from that in normal cells. Many tumor cells can produce ATP via the glycolytic pathway, even in a low-oxygen (20%) environment—referred to as aerobic glycolysis or the Warburg effect—and this is the most characteristic metabolic change that occurs in tumor cells. The synthesis of cellular macromolecules such as fats, nucleic acids, or amino acids, which are required for the proliferation of tumor cells, often branch off from the glycolytic pathway. In particular, the oncogenes MYC, AKT, and HIF-1α are known to activate glycolysis by regulating the expression of glucose transporters and glycolytic enzymes [1].
p53 regulates glycolysis via multiple mechanisms. Glucose transport is the first rate-limiting step in glucose metabolism. Glucose is transported into cells by glucose transporters GLUT1 and GLUT4, which are repressed at the transcriptional level by p53 [10]. Furthermore, p53 induces the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR) [11], which controls glycolysis by reducing intracellular fructose-2,6-bisphosphate levels and upregulating the pentose phosphate pathway (PPP). TIGAR is similar in structure to the glycolytic enzyme phosphofructokinase-2/fructose-2,6-bisphosphatase (PFK-2/FBPase-2). TIGAR functions to degrade fructose-2,6-bisphosphate (F-2,6-BP), which is a powerful allosteric activator of phosphofructokinase-1 (PFK-1). PFK-1 catalyzes the conversion of fructose-6-phosphate (F-6-P) to fructose-1,6-bisphosphonate (F-1,6-BP). F-2,6-BP also inhibits fructose-1,6-bisphosphatase and converts F-1,6-BP to F-6-P. By reducing F-2,6-BP levels and limiting the activity of PFK-1, TIGAR eventually contributes to reduce the rate of glycolysis [12]. p53 also regulates glycolysis by reducing levels of a glycolysis enzyme, phosphoglycerate mutase (PGM) [13]. Pyruvate dehydrogenase (PDH) connects glycolysis and the tricarboxylic acid (TCA) cycle. When PDH activity is increased, the conversion of pyruvate into acetyl-CoA is promoted, then TCA cycle and mitochondrial respiration is enhanced. PDH is negatively regulated by pyruvate dehydrogenase kinase isoform 2 (PDK2), and p53 further inhibits the expression of PDK2 [2, 14]. p53 also induces expression of the Parkin gene, which is associated with Parkinson’s disease, to downregulate glycolysis [15]. p53 inhibits expression of monocarboxylate transporter 1 (MCT1), a lactate transporter, inhibiting lactate uptake, which is produced via glycolysis under low-oxygen conditions. p53 thereby inhibits the shift in ATP production from mitochondrial oxidative phosphorylation to glycolysis [16].
The Akt/mTOR and NF-κB signaling pathways are often activated and upregulate glycolysis in many cancers, whereas p53 downregulates these pathways and inhibits glycolysis. p53 induces IGF-BP3 and PTEN expression, downregulates Akt signaling, and inhibits mTOR signaling by inducing AMPKβ, TSC2, Sestrin 1 and 2, and REDD1 expression [17]. p53 also inhibits glucose uptake and glycolysis by inhibiting GLUT3 expression via the downregulation of NF-κB signaling [18].
The PPP begins with glucose-6-phosphate (G6P) from the glycolytic pathway, resulting in NADPH and ribose-5-phosphate production, which is necessary for nucleic acid synthesis and this pathway is often activated in cancers. p53 negatively regulates PPP to inhibit production of NADPH and ribose-5-phosphate, which are required for tumor cell growth. Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme that catalyzes the first step of the PPP, and p53 suppresses G6PD through direct binding and reduces the level of NADPH by compromising the flow of G6P from glycolysis to the PPP [19].
In addition to its role negatively regulating glycolysis and the PPP, p53 plays an important role in mitochondrial oxidative phosphorylation and quality control. We previously reported that p53 induces expression of glutaminase 2 (GLS2), which hydrolyzes glutamine to glutamate. Glutamate is converted to α-ketoglutaric acid (α-KG), an important substrate in the TCA cycle. GLS2 upregulates the TCA cycle and oxidative phosphorylation by increasing α-KG levels [20]. In addition to suppression of glycolysis as mentioned previously, Parkin increases expression of pyruvate dehydrogenase E1α1, an important component of pyruvate dehydrogenase which catalyzes the conversion of pyruvate to acetyl-CoA, and in turn, upregulates the TCA cycle. Thus, Parkin contributes to the function of p53 in energy metabolism [15].
p53 activities are regulated via protein levels, coactivator or corepressor recruitment, and a wide variety of posttranslational modifications. One intriguing finding is the importance of lysine modification in controlling the p53 response and its metabolic regulation. The mutation of three lysine residues in mouse p53 (K117, K161 and K162, equivalent to K120, K164 and Q165 in human p53, respectively) abolished the ability of p53 to induce cell cycle arrest, apoptosis and senescence, though p53-3KR retained the ability to induce the metabolic target genes such as GLS2 and TIGAR. p53-3KR also inhibited the expression of GLUT3 and keeps the ability to suppress both glucose uptake and glycolysis. p53-3KR keeps the capacity to reduce ROS levels [21]. The association of metabolic regulation and p53 posttranslational modifications has not been fully examined so far. Future studies will be necessary to elucidate the effect of the posttranslational modifications on the selection of p53 multifaceted functions including metabolic regulation, via differentially transactivating p53 targets.
Malic enzymes (ME) are involved in the conversion of malate, an intermediate substrate within the TCA cycle, to pyruvate. MEs are also involved in the production of NADPH. Jiang et al. reported that p53 inhibits the expression of ME isoforms 1 and 2 (ME1 and ME2, respectively) [22]. p53 further controls mitochondrial DNA copy numbers and DNA synthesis via p53-controlled ribonucleotide reductase (p53R2) [23], and plays a role in mitochondrial quality control by repairing or degrading defective mitochondria via the induction of mitochondria-eating protein (Mieap) expression [24]. p53 is involved in the formation and functioning of mitochondrial complex I in the respiratory chain and induces the expression of apoptosis-inducing factor (AIF), an important factor in oxidative phosphorylation [25]. p53 also increases levels of cytochrome C oxidase 2 (SCO2), which plays an important role within mitochondrial complex IV [26].
Recently, we reported the significance of dihydropyrimidinase-like 4 (DPYSL4), a novel p53 inducible regulator of ATP production. DPYSL4 is present within mitochondrial complexes I, III, and IV, contributing to cellular energy production via the regulation of oxidative phosphorylation and has been shown to suppress cancer cell proliferation and invasion [27]. In summary, p53 negatively regulates glycolysis via the transcriptional regulation of numerous downstream genes involved in cellular metabolism (Fig. 2).

p53 and glucose metabolism
p53 regulates glycolysis by inducing TP53-induced glycolysis and apoptosis regulator (TIGAR) or Parkin expression; by inhibiting GLUT1, 3, and 4, phosphoglycerate mutase (PGM), pyruvate dehydrogenase kinase isoform 2 (PDK2), and monocarboxylate transporter 1 (MCT1) expression; and by inhibiting Akt/mTOR signaling. p53 induces synthesis of cytochrome C oxidase 2 (SCO2), apoptosis-inducing factor (AIF), p53R2, mitochondria-eating protein (Mieap), Parkin, glutaminase 2 (GLS2), and dihydropyrimidinase-like 4 (DPYSL4) expression; inhibits malic enzyme ME1 and ME2 expression; and promotes oxidative phosphorylation. p53 also inhibits glucose-6-phosphate dehydrogenase (G6PD) expression and the pentose phosphate pathway (PPP).
The fat composition in cancerous tissues is known to be different from that in normal tissues. It has become increasingly clear that various types of lipids contribute to the growth and development of cancer cells. Certain lipid metabolic pathways are also unique to cancers. For example, normal cells utilize extracellular fatty acids, whereas cancer cells often activate de novo fatty acid synthesis [28]. Such de novo synthesized fatty acids contribute to cancer cell survival and growth via lipid synthesis, membrane formation, energy production via β-oxidation, and regulation of signaling pathways. Expression of enzymes that participate in de novo fatty acid synthesis is selectively increased in several cancers, and inhibition of these enzymes can induce cancer-selective apoptosis and suppress proliferation. Molecules that regulate de novo fatty acid synthesis are considered highly promising targets for cancer treatment. NADPH is required for de novo fatty acid synthesis, which is also produced through glycolysis-PPP in cancer cells. In addition, acetyl-CoA, a key intermediate metabolite in the TCA cycle and a product of glycolysis, is also the initial substrate of fatty acid and cholesterol synthesis in lipid metabolism. p53 is involved in lipid metabolism via these two pathways.
Statins have been reported to be associated with reduced mortality of various types of cancers [29]. A recent study by Moon S.H. et al., revealed that p53 downregulates the mevalonate pathway. They identified that ABCA1 is a p53 target gene which inhibits SREBP-2 maturation under low-sterol conditions. ABCA1 contributes to suppression of liver tumorigenesis. Furthermore, they explored the gene expression during the initial stages of tumorigenesis using murine model of liver cancer. Intriguingly, they found that transcriptional upregulation of Abca1 accompanied by repression of mevalonate pathway genes even before the onset of apparent tumor [30].
Pantothenate kinase 1 (PANK1) catalyzes the rate-limiting step of CoA synthesis and has been identified as a target of p53 [31]. PANK1 does not have cell cycle arrest or apoptosis-inducing functions, but PANK1 is induced by DNA damage in a p53-dependent manner. Under glucose-starved conditions, PANK1 expression is maintained in the presence of p53, suggesting that p53 is required to maintain PANK1 expression in energy-depleted states.
Acetyl-CoA, the final product of fatty acid oxidation, is also an inhibitor of pyruvate dehydrogenase, which is the enzyme required for the final glycolysis step. Furthermore, acetyl-CoA is used in the TCA cycle to synthesize citrate, which inhibits a glycolytic enzyme, phosphofructokinase. Thus, p53 is thought to inhibit the progression of cancer by shifting lipid metabolism toward catabolism. p53 promotes catabolism of fatty acids while simultaneously acting to inhibit fatty acid synthesis. Fatty acid synthesis is dependent on the NADPH produced in the PPP. As noted above, p53 inhibits the function of G6PD and reduces NADPH levels by restricting the flow from glycolysis into the PPP [19].
There is significant crosstalk within the lipid metabolism regulatory system. Sterol regulatory element-binding protein (SREBP) refers to a family of transcription factors that regulate cholesterol, fatty acid, and triglyceride synthesis. The isoform SREBP1c activates expression of enzymes involved in fatty acid synthesis, such as fatty acid synthase (FASN) and ATP citrate lyase (ACLY). These enzymes are responsible for the increased fatty acid synthesis observed in many types of cancers [32, 33]. p53 decreases the expression of FASN and ACLY through inhibition of SREBP1c [34]. The mTOR pathway is also often activated in cancers. This pathway acts to enhance fatty acid synthesis and is suppressed by p53 [35]. As noted above, p53 inhibits the expression of ME1 and ME2. Malate is converted to pyruvate by MEs, and the NADPH produced in this process is used in fatty acid synthesis. Cellular aging and tumor reduction associated with p53 activation has been observed upon knockdown of ME. Furthermore, serum triglyceride levels and overall lipid accumulation are reduced in response to reduced intracellular NADPH [22]. These results show that p53 exerts cancer-suppressive functions via MEs and associated feedback mechanisms (Fig. 3).

Regulation of fatty acid metabolism by p53
p53 promotes the catabolism of fatty acids while simultaneously inhibiting fatty acid synthesis.
There has been recent interest in the influence of sphingolipid metabolism on tumorigenesis due to reports of sphingolipid metabolism disorders in various cancers [36, 37]. Sphingolipids are a group of lipids that have sphingoid bases within their structures. Sphingolipid molecules include sphingosine and ceramide, which control cell growth and differentiation, cell cycle, apoptosis, and cellular aging. Sphingosine kinase 1 (SK1), the main enzyme in sphingolipid metabolism, phosphorylates sphingosine to produce sphingosine-1-phosphate (S1P). SK1 is important for maintaining the balance between S1P which has cell proliferative functions and sphingosine and ceramide which have apoptosis-inducing effects. SK1 is reportedly activated in various cancers [38]. Heffernan-Stroud et al. found SK1 is proteolyzed via activation of caspase-2 in a p53-dependent manner [39]. In p53-knockout mice, SK1 expression is increased, resulting in an increase in S1P and a decrease in ceramide levels. Tumor formation induced by p53 gene deletion is reversed by SK1 gene deletion, thus indicating the importance of sphingolipid metabolism and p53 regulation in tumorigenesis. Ceramide is an intermediate substrate of sphingolipid metabolism that has cell cycle arrest functions and can induce apoptosis. Ceramide synthase 6 (CerS6), a ceramide synthesis enzyme, is also a p53-dependent downstream gene [40]. There are other pathways for ceramide synthesis, such as those catalyzed by sphingomyelinase from sphingomyelin, which is the most abundant sphingolipid in cells (Fig. 4), but their detailed relationship with p53 remains to be elucidated in future studies.

Sphingolipid metabolism
Sphingolipids consist of substances such as sphingosine, sphingosine-1-phosphate (S1P), ceramide, and sphingomyelin. Sphingosine kinase 1 (SK1), which produces S1P by phosphorylation of sphingosine, is important for maintaining the balance between S1P—which has cell proliferative effects—and sphingosine or ceramide—which have apoptosis-inducing effects.
It has become increasingly evident that dramatic changes in amino acid metabolism which are closely related to glucose metabolism in cancer cells. Amino acids not only function as protein building blocks but also serve as basic components of other macromolecules such as lipids and sugars. In rapidly proliferating cells (e.g., cancer cells), energy metabolism becomes reliant on amino acids after glucose depletion. In particular, glutamine becomes an important energy source under low-glucose conditions. Glutamine is converted to glutamate by glutaminase. Glutamate is then converted to α-KG by glutamine dehydrogenase, and α-KG then activates ATP synthesis by entering the TCA cycle. The overall process by which glutamine is decomposed into lactate via α-KG is called glutaminolysis. p53 regulates glutamine metabolism through the aforementioned induction of GLS2 expression [20]. p53-activated GLS2 makes cellular energy production possible under glucose-deprivation conditions, and thus plays an important role in cell survival and growth (Fig. 5). Glutamate is also a substrate for the synthesis of the reduced form of glutathione (GSH), which acts antagonistically to limit intracellular and extracellular oxidative stress, making it important in cell survival (Figs. 5, 6).

Intracellular metabolic regulation by p53
p53 regulates the metabolic pathway by controlling the transcriptional activity of intracellular metabolic enzymes. Glutaminase 2 (GLS2), which is involved in glutamine metabolism, increases α-KG levels, which in turn promotes the tricarboxylic acid (TCA) cycle and oxidative phosphorylation and promotes antioxidative effects via glutathione (GSH) synthesis.

Mechanism of oxidative stress response regulation by p53
Under mild metabolic stress, p53 exerts antioxidative effects. Overproduction of reactive oxygen species (ROS) leads to the induction of apoptosis via p53 activation. GSH, glutathione; PPP, pentose phosphate pathway.
Cancer cells tend to be exposed to nutrient-deprived conditions, but possess the means to survive. Glutamine is recognized as the most abundant amino acid in the body. As described above, glutamine is not only involved in energy production via the TCA cycle and antioxidant defense via glutathione synthesis but also contributes to nucleotide and amino acid synthesis. Tajan et al. identified that p53 contributes to the adaptation of cancer cells to glutamine starvation via the transactivation of SLC1A3, an aspartate/glutamate transporter. Aspartate metabolism is important under glutamine starvation conditions [41]. SLC1A3 is localized to the inner mitochondrial membrane, and is involved in importing extracellular aspartate and functions in the malate/aspartate shuttle (MAS) as an aspartate-glutamate carrier [42, 43] to provide reducing equivalents and support electron transport chain (ETC) (Fig. 7). In addition to SLC1A3, there are two canonical mitochondrial transporters, AGC1 (Aralar; SLC25A12) and AGC2 (Citrin; SLC25A13). Neither of them were clearly influenced by p53 though AGC2 was slightly induced by glutamine starvation in p53 wild-type cells. Thus, p53 functions to protect cells from metabolic stress such as nutrient deprivation.
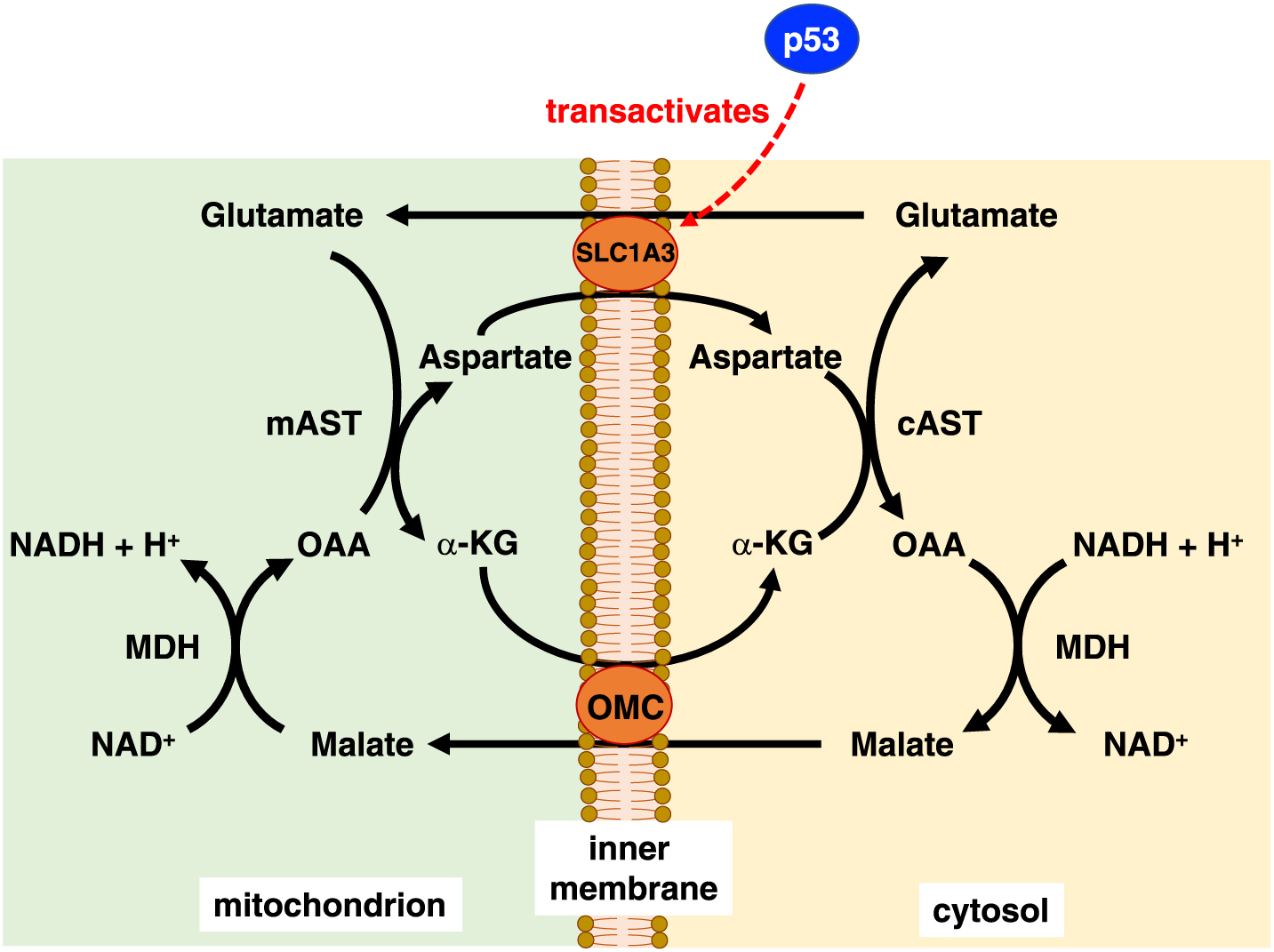
Schematic of the malate-aspartate shuttle
p53 contributes to the adaptation of cancer cells to glutamine starvation through transactivation of SLC1A3. SLC1A3 is localized to the inner mitochondrial membrane and functions as a glutamate transporter in the malate/aspartate shuttle, which transfers reducing equivalents between the cytosol and mitochondria and thus supports the electron transport chain. AST, aspartate aminotransferase; α-KG, α-ketoglutarate; MDH, malate dehydrogenase; OAA, oxaloacetate; OMC, oxoglutarate-malate carrier.
In recent years, serine has been recognized as incredibly important in tumor cell survival and proliferation. Serine activates the glycolytic enzyme pyruvate kinase M2 (PKM2) [44] (Fig. 8). When the activation of PKM2 is suppressed due to serine starvation, glycolysis is inhibited and the flow into the TCA cycle increases. p53 not only inhibits cell proliferation but also promotes serine synthesis and causes synthesis of GSH, thereby contributes to overall cell survival [45]. The serine synthetic pathway plays an important role in providing α-KG, an intermediate of TCA cycle that is necessary for cancer cell growth. Serine is also reported to act on the methionine pathway and to affect regulation of DNA/RNA methylation [46] (Fig. 8). p53 can inhibit proliferation or induce cell death, but also help cell survival by induction of DNA repair pathway or adaptation to environmental changes. As a tumor suppressor, it seemingly sounds contradictory that wild-type p53 contributes to cell survival, but there is growing understanding that p53 could support adaptation and survival of cells in response to nutrient depletion. In consequence of exerting tumor suppressor function, p53 could promote the cell survival depending on the context, and these functions may contribute to maintain homeostasis beyond cancer development [41].

Regulation of cellular metabolism by p53 in response to serine depletion
Glucose is metabolized to 3-phosphoglycerate (3-PG) in the glycolytic pathway. Serine enters the serine biosynthesis pathway, which branches off from glycolysis. Serine activates pyruvate kinase M2 (PKM2), which is a glycolytic enzyme. In contrast, during serine depletion, serine biosynthesis is activated in response to decreased levels of PKM2. Synthesized serine is converted to glycine and causes cell cycle arrest, inhibits purine synthesis, and increases production of the reduced form of glutathione (GSH) in a p53/p21-dependent manner. GSH suppresses reactive oxygen species (ROS) that are produced in the tricarboxylic acid (TCA) cycle.
Thus far, we have summarized the mechanisms by which p53 maintains order in cellular metabolism. Disorderly cellular activity or energy production leads to excessive metabolic stress, including the production of reactive oxygen species (ROS), and may lead to changes in the intracellular environment or even apoptosis. Excess ROS generated in cells activates apoptosis-inducing factors, such as p53-induced gene 3 (PIG3) [47], proline oxidase (POX) [48], BAX, PUMA [49], and p66Shc [50]. Thus, excess ROS activates p53 and induces apoptosis by promoting the production and release of ROS from mitochondria via positive feedback cycle. However, under physiological condition, antioxidative factors such as Sestrin family are transactivated downstream of p53 and play an important role in antioxidative effects [51]. p53 target genes Sesn1 and Sesn2 inhibit mTOR signaling via activating AMPK [51]. As noted above, GLS2 also exerts antioxidative effects via GSH synthesis [20]. Sablina et al., explored the antioxidant function of p53 in cancer cells. They found that p53 might have opposing roles in the regulation of ROS depending on the degree and the duration of the stress. The cells expressing exogenous p53 showed decreased ROS levels with short duration, but there was a substantial increase in intracellular ROS levels with prolonged overexpression of p53. Furthermore, whereas the cells treated with mild stress showed decreased ROS, p53-positive cells which treated with severe stress showed increased ROS levels, resulting in apoptosis [52]. Therefore, p53 has both positive and negative effects on regulation of ROS. p53 functions to maintain homeostasis under basal or low-stress conditions while also eliminating irreversibly damaged cells and may determine the cell fate by selecting specific target gene(s) depending on the degree or type of cellular stress (Fig. 6). Both of these response modes would help to prevent the progression of malignancy.
In a murine model encoding p53 Lys to Arg mutations in three acetylated loci important for apoptosis induction and cell cycle arrest, such effects were decreased although energy metabolism regulation and antioxidative effects were maintained. These results suggest p53 may exert tumor-suppressive functions in the early phases of tumorigenesis by controlling intracellular metabolism [21, 53]. The tumor suppressor functions of p53 can be considered a safety net that functions in a multilateral and multistep manner.
Most mutations in p53 occur as missense mutations. Mutant p53 not only loses its function but can also acquire new functions (i.e., “gain-of-function” mutations) such as cell proliferation, angiogenesis, metastasis, and chemotherapy resistance that lead to the promotion of cancer. Most mutations in mutant p53 are concentrated in DNA-binding domains, including hotspots such as R175, G245, R248, R249, R273, and R282. In a murine model of Li-Fraumeni syndrome, mice that express p53 with R172H or R270H mutations (equivalent to R175H and R273H, respectively, in humans) have a higher tumorigenic potential and metastatic capacity, in consequence, promotes cancer progression compared to p53-knockout mice [54, 55].
The functions of mutant p53, including changes in cellular metabolism, vary according to the locus of the mutation (Fig. 9). The mevalonate pathway, which is involved in cholesterol synthesis, is related to many aspects of tumor formation such as growth, survival, invasion, and metastasis. Mutant p53 R280K and R273H induces the expression and activation of genes related to the mevalonate pathway via binding to the transcription factor SREBP, then leads to the degradation of mammary gland structures [56]. Mutant p53 R172H promotes translocation of GLUT1 to the cell membrane through activation of RhoA/ROCK (Rho-associated protein kinase) signaling, thereby inducing glycolysis while promoting tumor formation [57]. Furthermore, under conditions of metabolic stress including glucose-starvation or low-oxygen conditions, mutant p53 promotes lipid production, glycolysis, and tumor growth by inhibiting AMPK activity via binding to a subunit of AMPKα [58]. These findings show that p53 mutations are important for initiating metabolic changes in cancer, and increased understanding of these mechanisms may lead to the discovery and development of novel treatments focused on cellular metabolism.
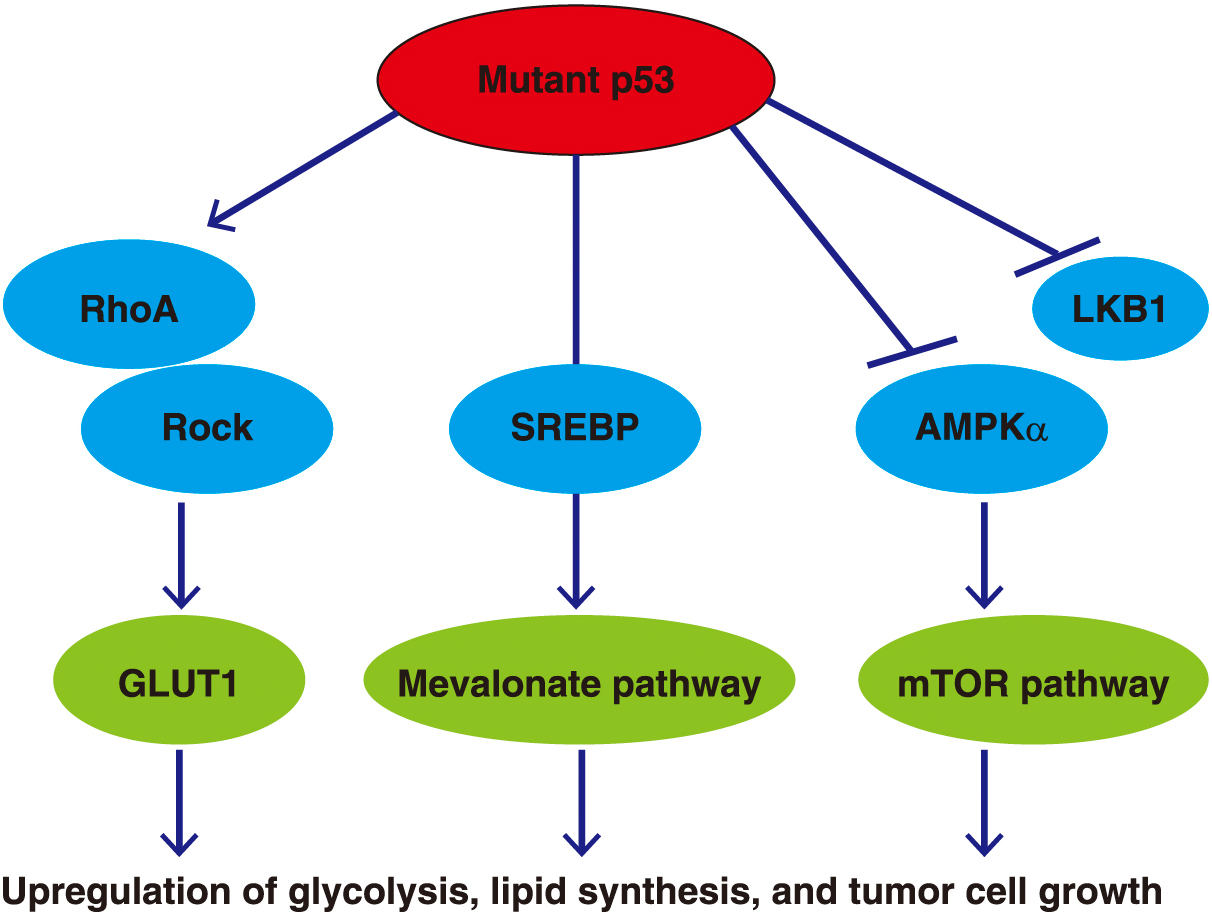
Mutant p53 and intracellular metabolism
Gain-of-function p53 mutations result in the upregulation of glycolysis, lipid biosynthesis, and tumor cell growth via their effects on intracellular metabolic enzymes.
Since it was firstly reported in 1979, the tumor suppressor gene p53 has been found to play a role in not only cell cycle arrest or apoptosis but also in various other physiological functions, including cellular aging and stem cell regulation. This regulation involves epigenome, metabolic homeostasis, mitochondrial function and ROS/energy control as well. Therefore, the role of p53 is focused in a lot of fields including diabetes, obesity and cardiovascular disorders, and recent studies have uncovered that intracellular and extracellular metabolic environments and energy states are closely associated with p53-related transcriptional environments and epigenomic regulatory functions in order to maintain cellular homeostasis or adapt to new environments. Following the discovery of oncometabolites and their subsequent functional analysis, it has also been established that 2-hydroxyglutarate and fumarate metabolites are closely associated with tumor formation, and that both α-KG and S-adenosyl methionine interfere with genomic activity by controlling the epigenetic regulatory factors. In addition to p53, transcription factors and intra/extracellular metabolic changes form a network using these chemical modifications and systems, and then the gene expressions are totally regulated with altered the genome structure. This system is closely related to stem cell regulation or aging signals, and it can be easily imagined that abnormal functioning of this system would lead to the pathophysiology of cancer and metabolic diseases. In the future, further advancements in single-cell analysis and genome editing technologies, together with multi-omics analyses and innovation in big data analysis technology, will provide in-depth understanding of single gene, single molecule, and single cell as a part of dynamic system that broadly connects cells to the individual. From this framework, we expect new breakthroughs in the understanding of p53-related mechanisms of metabolic control and the molecular pathogenesis of cancer and other lifestyle-related diseases.
None of the authors have any potential conflicts of interest associated with this review article.