Abstract
McCune–Albright syndrome (MAS) is a rare disorder. MAS is classically defined by the occurrence of fibrous dysplasia, café-au-lait skin macules, and precocious puberty. In addition to precocious puberty, other hyperfunctioning endocrinopathies may occur. We evaluated hypothalamic–pituitary–adrenal function in two cases of typical MAS associated with fibrous dysplasia and growth hormone excess. Pituitary adenoma or hyperplasia was not detected by magnetic resonance imaging. Hormonal data showed normal or low cortisol levels, despite high ACTH levels in the blood. A high ratio of circulating ACTH to cortisol was found in the two cases. Insulin tolerance and CRH tests showed hyper-responses of ACTH and an insufficient increase in cortisol levels. No involvement of 11β-HSD1 by GH excess was suggested because basal levels of ACTH and cortisol showed no changes, even after therapy for acromegaly by somatostatin analogues. Patients with Cushing’s disease cases of pituitary macroadenoma can have high circulating ACTH precursor levels, and elevated ACTH precursors have been observed in ectopic ACTH syndrome. Autonomous cortisol excess was excluded by the level of midnight cortisol and the level of cortisol after a low-dose dexamethasone suppression test in the two cases. Finally, the gel filtration profiles of immunoreactive ACTH contents showed the presence of aberrant ACTH precursors. To the best of our knowledge, there have been no reports of MAS associated with aberrant ACTH precursors. Our findings in these cases emphasize that attention should be to secretion of inactive ACTH precursors in MAS.
MCCUNE–ALBRIGHT SYNDROME (MAS) is a rare disorder, which has an estimated prevalence of between 1:100,000 and 1:1,000,000 [1]. MAS is caused by an activating postzygotic somatic mutation in the α subunit of the stimulatory G protein gene (GNAS). Detection of mutation depends on the level of mosaicism in the tissue and the sensitivity of the technique. The detection rate is generally low in unaffected tissues. Thus, mutations in peripheral blood leukocytes cannot be detected by standard PCR-based Sanger sequencing. The diagnosis of MAS is established in individuals who have two or more typical clinical features. MAS is defined by occurrence of fibrous dysplasia (FD), café-au-lait skin macules, and endocrinopathies that result from hyperactivity of a variety of endocrine glands, including precocious puberty, hyperthyroidism, or excess growth hormone [1]. The clinical manifestations of MAS are variable and depend on the degree of mosaicism. The reason for this variability is presumably due to variability of mutation abundance among affected tissues.
MAS is sometimes reported to show hypercortisolism. Cushing’s syndrome is a potentially fatal feature of MAS. ACTH-independent macronodular adrenal hyperplasia is rarely accompanied by MAS [2]. There have been no reports of ACTH excess, such as Cushing’s disease and ectopic ACTH syndrome, in MAS. Patients with pituitary corticotroph macroadenoma can have high circulating levels of ACTH precursors [3, 4]. ACTH precursors are also greatly elevated in patients with ectopic ACTH syndrome [5-7]. Aberrant pro-opiomelanocortin (POMC) processing is considered in patients with pituitary corticotroph macroadenoma or with ectopic ACTH syndrome.
We report normal or low cortisol levels, despite high ACTH levels in two cases of MAS. Hypothalamic–pituitary–adrenal (HPA) function was evaluated by endocrinological examinations. The gel filtration profiles of immunoreactive ACTH contents showed the presence of aberrant ACTH precursors.
Measurement of ACTH and Cortisol Levels
Plasma ACTH and serum cortisol levels were assessed using electrochemiluminescence immunoassays (Eclusys ACTHTM and Eclusys Cortisol IITM, respectively; Roche Diagnostics K.K., Tokyo, Japan).
Gel Exclusion Chromatography
Gel exclusion chromatography was performed as described previously [8].
Case Presentation
Case 1
A 42-year-old man was referred for further evaluation of MAS. He showed a height of 179.2 cm, a weight of 69.6 kg, and body mass index of 21.7 kg/m2. Blood pressure was 121/68 mmHg without antihypertensive agents. He showed craniofacial deformities (Fig. 1A) and suffered from multiple bone fractures. He was diagnosed with FD at the age of 23 years. Café-au-lait macules were found on his back. He had slightly acromegaloid features with supraorbital ridges and showed external occipital protuberance (Fig. 1A). X-ray imaging showed flower cabbage-like deformities of the distal phalanx of the fingers (image not shown). He showed no cushingoid features. He had a huge goiter and a 4-year history of hyperthyroidism under treatment with methimazole. The diagnosis of a toxic multinodular goiter was confirmed by technetium-99m pertechnetate thyroid scintigraphy (images not shown).
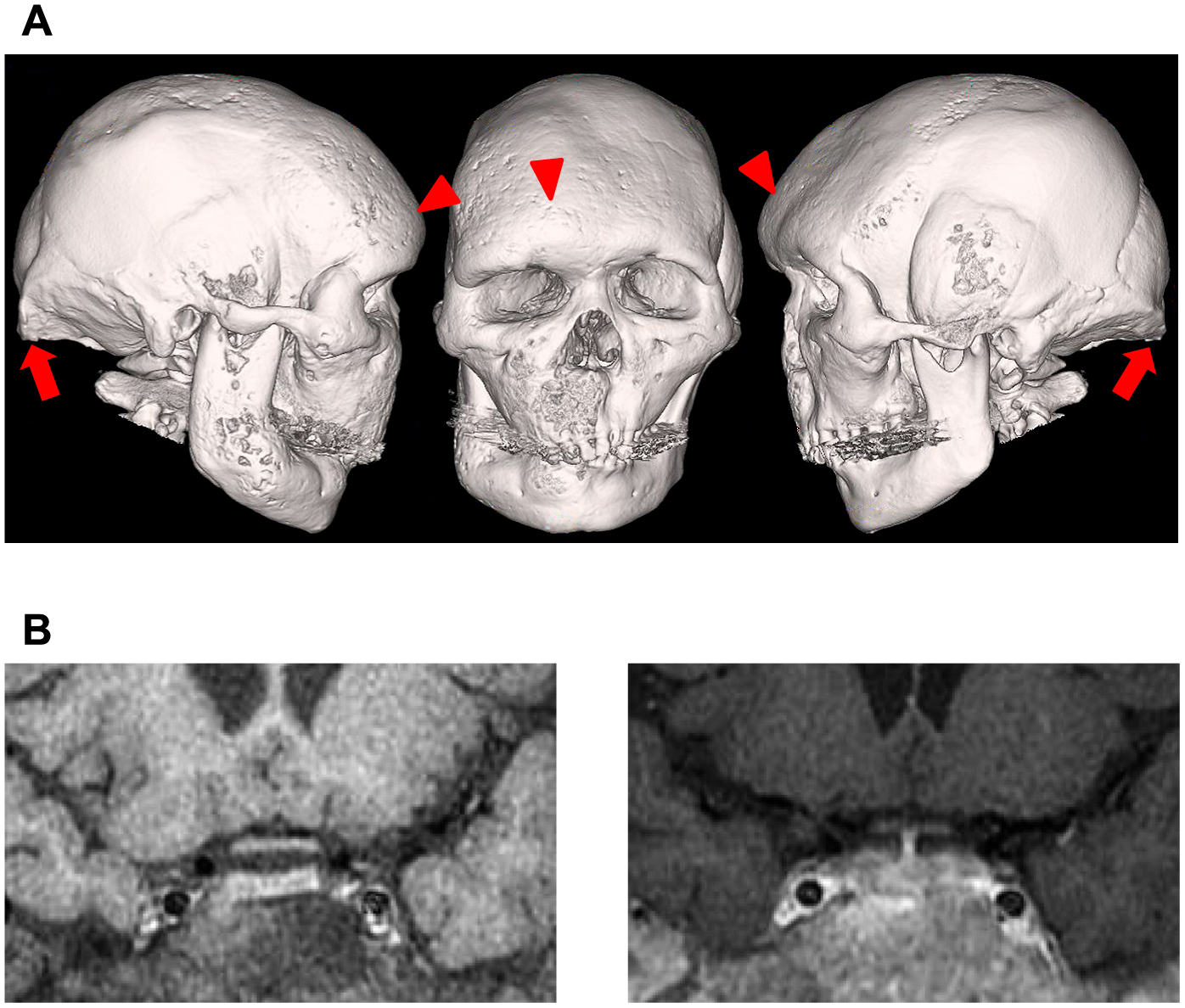
A complete blood count, serum electrolytes, and renal and liver tests were normal. He also showed normal glucose tolerance. Elevated alkaline phosphatase levels and markers of bone metabolism were observed because of FD (data not shown). Pituitary adenoma or hyperplasia was not detected by magnetic resonance imaging (MRI) (Fig. 1B). However, the diagnosis of GH excess was confirmed by no suppression of serum GH levels by a 75-g oral glucose tolerance test (nadir GH: 2.34 ng/mL) and an elevated serum insulin-like growth factor-I level (307 ng/mL; normal range: 92–257 ng/mL). Serum GH levels were paradoxically increased after stimulation with TRH, but not with LHRH and CRH (data not shown). Serum GH levels were decreased after subcutaneous injection of 100 mg octreotide. Basal serum PRL levels were 62.5–68.9 ng/mL. A TRH test showed a suboptimal response of PRL (peak level: 90.8 ng/mL). PRL levels were decreased after oral administration of bromocriptine, but not subcutaneous injection of 100 mg octreotide (data not shown).
Basal ACTH and cortisol levels in blood were 40.6–63.4 pg/mL and 8.0–10.5 μg/dL, respectively. The ratio of circulating ACTH to cortisol was 5.1 to 6.0. The urinary free cortisol level was 53 μg/day. Autonomous cortisol excess was excluded by the level of midnight cortisol (1.2 μg/dL) and the level of cortisol (0.2 μg/dL) after a low-dose (1 mg) dexamethasone suppression test. A rapid ACTH stimulation test showed a suboptimal response (Fig. 2A). A repeated ACTH simulation test showed a sufficient increase in urinary free cortisol levels (up to 3,640 μg/day). An insulin tolerance test showed hyper-response of ACTH and an insufficient increase in cortisol levels (Fig. 2B). A CRH test showed a normal response of ACTH and an insufficient response in cortisol levels (Fig. 2C). Serum levels of free triiodothyronine (2.64 pg/mL), free thyroxine (0.40 ng/dL), and TSH (1.64 μU/mL) were within normal limits under treatment with 5 mg of methimazole. Basal TSH, LH, and FSH levels in serum were within the normal range. TRH and LHRH tests showed a normal response of these hormones (data not shown). The patient was treated with monthly subcutaneous lanreotide injection and then GH excess was well controlled.

A 32-year-old man was admitted for further evaluation of the HPA axis on receiving the results of Case 1. MAS associated with gigantism was diagnosed at the Pediatrics Department at the age of 5 years. GH excess had been treated with a dopamine receptor agonist for 13 years. Transition from pediatric to adult care was performed at the age of 18 years. Treatment of GH excess was then followed by monthly octreotide depot for 14 years and was well controlled (serum insulin-like growth factor-I levels were approximately 200 ng/mL; normal range of a 32-year-old man: 105–292 ng/mL). His height was 178.1 cm, weight was 67.0 kg, and body mass index was 21.1 kg/m2. Blood pressure was 108/68 mmHg without antihypertensive agents. He showed craniofacial deformities (Fig. 3A). He had no acromegaloid features and no cushingoid features. Café-au-lait macules were observed from the left flank to the back. Pituitary adenoma or hyperplasia was not detected by MRI (Fig. 3B).

A complete blood count, serum electrolytes, and renal and liver tests were within normal levels. Alkaline phosphatase levels and bone metabolism markers were elevated. Glycosylated hemoglobin A1c and fasting plasma glucose levels were 5.0% and 79 mg/dL, respectively. Basal ACTH and cortisol levels in blood were 35.5–73.1 pg/mL and 7.0–11.7 μg/dL, respectively. The ratio of circulating ACTH to cortisol was 5.1 to 6.2. The urinary free cortisol level was 61 μg/day. A rapid ACTH stimulation test revealed a normal response (Fig. 2D). Repeated ACTH simulation tests also showed a sufficient increase in urinary free cortisol levels (up to 3,690 μg/day). An insulin tolerance test showed a normal response of ACTH and an insufficient increase in cortisol levels (Fig. 2E). A CRH test showed a normal response of ACTH and an insufficient response of cortisol levels (Fig. 2F). Autonomous cortisol excess was excluded by the level of cortisol (<0.2 μg/dL) after a low-dose (0.5 mg) dexamethasone suppression test. Basal serum TSH, PRL, LH, and FSH levels were within the normal range. TRH and LHRH tests showed a normal response of these hormones (data not shown).
On the basis of the results of the two cases, the presence of high-molecular ACTH was suspected. Gel exclusion chromatography was then performed. POMC and pro-ACTH were detected and the aberrant ACTH/total ACTH ratio was 42% in both cases (Fig. 4).
Discussion
Despite higher ACTH levels, normal or lower levels of cortisol were observed in both of our patients. At first, primary adrenal insufficiency was excluded by endocrinological examinations. Both patients had GH excess. Therefore, we next considered the possibility of 11β-hydroxysteroid dehydrogenase type 1 (HSD1) activity because 11β-HSD1 can be inhibited by GH excess. In fact, GH is an important regulator of 11β-HSD1 activity, which is evaluated as the ratio of cortisol/cortisone [9-12]. Unfortunately, serum and urinary cortisone levels were not evaluated in our patients. However, basal levels of ACTH and cortisol showed no changes, even after therapy for acromegaly by somatostatin analogues. This finding suggests no involvement of 11β-HSD1 by GH excess in our cases.
Hypercortisolism due to ACTH-independent macronodular adrenal hyperplasia has been reported in MAS [2]. However, there have been no reports of ACTH excess, such as Cushing’s disease and ectopic ACTH syndrome, in MAS. Concomitant GH- and ACTH-producing adenomas have been rarely reported [13-15, 16], resulting in acromegaly and overt or subclinical Cushing’s disease. Additionally, up to 25% of GH-producing adenomas concomitantly produce PRL. However, such findings of a pituitary tumor or hyperplasia were not observed in either of our cases. In our cases, the possibility of Cushing’s disease or subclinical Cushing’s disease was also excluded because of sufficient suppression of plasma cortisol levels by a low-dose overnight dexamethasone suppression test.
The presence of high molecular ACTH was then suspected in our cases because the ratio of circulating ACTH to cortisol was considered to be high. In fact, an ACTH/cortisol ratio of 5.1 to 6.2 was found in our cases. The ratio of circulating ACTH to cortisol is higher in Cushing’s disease cases of macroadenoma compared with microadenoma [3, 17]. This could be due to reduced processing of POMC to ACTH in large pituitary tumors. Similar cases have been observed in ectopic ACTH syndrome [5-7]. While the ACTH to cortisol ratio showed between 2.4 and 2.6 and between 5.1 and 5.5 in cases of microadenoma and macroadenoma, respectively [17], a ratio >7 may be found in ACTH-producing ectopic pituitary adenoma [7]. The Eclusys ACTHTM was used for measurement of plasma ACTH levels in our cases. However, this assay fails to distinguish among POMC, pro-ACTH, and authentic ACTH. Gel filtration chromatography is able to distinguish these hormones, and it shows the presence of aberrant ACTH precursors.
Prohormone convertase (PC) 1/3 acts on the first cleavages of POMC to yield the N-terminus of POMC, ACTH, and β–lipotropin [18]. Increased levels of unprocessed POMC and a decreased ratio (relative to POMC) of processed POMC-derived peptides were found in PC1/3 knockdown and knockout human embryonic stem cell-derived neurons [19]. PC1/3 deficiency is associated with reduced processed ACTH secretion [19]. Most patients with PC1/3 deficiency suffer from early-onset obesity and hyperphagia, and approximately two thirds of these patients show reduced plasma ACTH levels, increased ACTH precursors, and almost normal cortisol levels [20]. In both of our cases, such types of Cushing’s syndrome and PC1/3 deficiency were excluded.
Peak serum cortisol levels >18 μg/dL by a rapid ACTH stimulation test or insulin tolerance test are considered to be normal HPA function. Serum cortisol levels >18 μg/dL and peak plasma ACTH levels >50 pg/mL, or peak plasma ACTH levels increase more than two-fold the basal level by the CRH test are considered to be normal function of the HPA axis [21-23]. According to these criteria, a rapid ACTH stimulation test showed suboptimal and sufficient responses in Cases 1 and 2, respectively. However, a repeated ACTH simulation test showed sufficient increases in urinary free cortisol in both cases. These results exclude the possibility of primary adrenal insufficiency [23]. Insulin tolerance and CRH tests showed hyper-responses of ACTH and an insufficient increase in cortisol levels. Therefore, these results suggested secondary adrenal insufficiency.
CRH is essential for ACTH under stress conditions, and it has an important role in processing of POMC [24]. Patients with impaired processing of ACTH show secondary hypocortisolism [25, 26]. These results suggest that endogenous CRH and ACTH in patients might have a low bioactivity. Indeed, the ACTH precursor (POMC+pro-ACTH)/total ACTH ratio was 42% in our cases. This result suggests that only 58% of plasma ACTH is authentic, and it can have normal bioactivity at its receptors, and stimulate cortisol secretion. However, elevated plasma ACTH levels, including ACTH precursors, might sufficiently maintain physiological levels of circulating cortisol.
Our patients visit our department monthly for treatment of GH excess. Treatment with hydrocortisone is considered for patients in special situations, such as sickness, operations, and drug interactions, and they are repeatedly educated to avoid life-threatening emergencies.
Some limitations of our cases should be taken into consideration. Around 10% of MAS have somatic activating (gain-of-function) variants at GNAS in the anterior pituitary diseases that can lead to GH excess; 81% of individuals with autonomous GH production also have hyperprolactinemia in MAS has been reported [1, 27]. However, there have been no reports of ACTH excess in MAS. Although GNAS mutations have been described in cortisol-producing adrenocortical diseases that can lead to Cushing’s syndrome, they have not been reported in Cushing’s disease. Because we were unable to obtain pituitary-derived tissue, we could not confirm the source of ACTH precursors, GH, and PRL in Case 1. Furthermore, assessment of expression levels or mutations of PC1/3, and evaluation of levels of tissue-specific processing of POMC were not investigated.
In conclusion, this is the first report of the presence of aberrant ACTH precursors, particularly POMC, in MAS. A high ratio of circulating ACTH to cortisol may suggest secretion of inactive ACTH precursors in MAS. Further investigations are required to determine whether GNAS mutations or other mechanisms are involved in the presence of aberrant ACTH precursors in MAS.
Acknowledgment
We thank Ellen Knapp, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Disclosure
None of the authors have any potential conflicts of interest associated with this research. Consent was obtained from the patients for publication of this case report.
References
- 1 Boyce AM, Florenzano P, Castro LF, Collins MT (2018) Fibrous dysplasia/McCune–Albright syndrome. In: Adam MP (ed) GeneReviews. University of Washington, Seattle, USA: internet.
- 2 Carney JA, Young WF, Stratakis CA (2011) Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune–Albright syndrome. Am J Surg Pathol 35: 1311–1326.
- 3 Gibson S, Ray DW, Crosby SR, Dornan TL, Jennings AM, et al. (1996) Impaired processing of proopiomelanocortin in corticotroph macroadenomas. J Clin Endocrinol Metab 81: 497–502.
- 4 Reincke M, Allolio B, Saeger W, Kaulen D, Winkelmann W (1987) A pituitary adenoma secreting high molecular weight adrenocorticotropin without evidence of Cushing’s disease. J Clin Endocrinol Metab 65: 1296–1300.
- 5 Stewart PM, Gibson S, Crosby SR, Penn R, Holder R, et al. (1994) ACTH precursors characterize the ectopic ACTH syndrome. Clin Endocrinol (Oxf) 40: 199–204.
- 6 Oliver RL, Davis JR, White A (2003) Characterisation of ACTH related peptides in ectopic Cushing’s syndrome. Pituitary 6: 119–126.
- 7 Suzuki J, Otsuka F, Ogura T, Kishida M, Takeda M, et al. (2004) An aberrant ACTH–producing ectopic pituitary adenoma in the sphenoid sinus. Endocr J 51: 97–103.
- 8 Morita K, Ogawa M, Kimura M, Okawa Y, Oki Y, et al. (2019) Falsely elevated plasma ACTH levels measured by the Elecsys assay related to heterophilic antibody in a case of secondary adrenocortical insufficiency. Endocr J 66: 563–569.
- 9 Moore JS, Monson JP, Kaltsas G, Putignano P, Wood PJ, et al. (1999) Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by growth hormone and insulin-like growth factor: in vivo and in vitro studies. J Clin Endocrinol Metab 84: 4172–4177.
- 10 Trainer PJ, Drake WM, Perry LA, Taylor NF, Besser GM, et al. (2001) Modulation of cortisol metabolism by the growth hormone receptor antagonist pegvisomant in patients with acromegaly. J Clin Endocrinol Metab 86: 2989–2992.
- 11 Giavoli C, Libé R, Corbetta S, Ferrante E, Lania A, et al. (2004) Effect of recombinant human growth hormone (GH) replacement on the hypothalamic-pituitary-adrenal axis in adult GH-deficient patients. J Clin Endocrinol Metab 89: 5397–5401.
- 12 Mondok A, Varga I, Glaz E, Szucs N, Tóth M, et al. (2009) 11beta-hydroxysteroid dehydrogenase activity in acromegalic patients with normal or impaired carbohydrate metabolism. Steroids 74: 725–729.
- 13 Horvath E, Kovacs K (2006) Pathology of acromegaly. Neuroendocrinology 83: 161–165.
- 14 Melmed S (2006) Medical progress: acromegaly. N Engl J Med 355: 2558–2573.
- 15 Oki K, Yamane K, Oda Y, Kamei N, Watanabe H, et al. (2009) Combined acromegaly and subclinical Cushing disease related to high–molecular–weight adrenocorticotropic hormone. J Neurosurg 110: 369–373.
- 16 Kageyama K, Nigawara T, Kamata Y, Terui K, Anzai J, et al. (2002) A multihormonal pituitary adenoma with growth hormone and adrenocorticotropic hormone production, causing acromegaly and Cushing disease. Am J Med Sci 324: 326–330.
- 17 Selvais P, Donckier J, Buysschaert M, Maiter D (1998) Cushing’s disease: a comparison of pituitary corticotroph microadenomas and macroadenomas. Eur J Endocrinol 138: 153–159.
- 18 Cawley NX, Li Z, Loh YP (2016) 60 YEARS OF POMC: Biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J Mol Endocrinol 56: T77–T97.
- 19 Wang L, Sui L, Panigrahi SK, Meece K, Xin Y, et al. (2017) PC1/3 Deficiency impacts pro-opiomelanocortin processing in human embryonic stem cell-derived hypothalamic neurons. Stem Cell Reports 8: 264–277.
- 20 Stijnen P, Ramos-Molina B, O’Rahilly S, Creemers JW (2016) PCSK1 mutations and human endocrinopathies: from obesity to gastrointestinal disorders. Endocr Rev 37: 347–371.
- 21 Maghnie M, Uga E, Temporini F, Di Iorgi N, Secco A, Tinelli C, et al. (2005) Evaluation of adrenal function in patients with growth hormone deficiency and hypothalamic-pituitary disorders: comparison between insulin-induced hypoglycemia, low-dose ACTH, standard ACTH and CRH stimulation tests. Eur J Endocrinol 152: 735–741.
- 22 Charmandari E, Nicolaides NC, Chrousos GP (2014) Adrenal insufficiency. Lancet 383: 2152–2167.
- 23 Yanase T, Tajima T, Katabami T, Iwasaki Y, Tanahashi Y, et al. (2016) Diagnosis and treatment of adrenal insufficiency including adrenal crisis: a Japan Endocrine Society clinical practice guideline [Opinion]. Endocr J 63: 765–784.
- 24 Fukuda Y, Kageyama K, Nigawara T, Kasagi Y, Suda T (2004) Effects of corticotropin-releasing hormone (CRH) on the synthesis and secretion of proopiomelanocortin-related peptides in the anterior pituitary: a study using CRH-deficient mice. Neurosci Lett 367: 201–204.
- 25 O’Rahilly S, Gray H, Humphreys PJ, Krook A, Polonsky KS, et al. (1995) Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med 333: 1386–1390.
- 26 Nussey SS, Soo SC, Gibson S, Gout I, White A, et al. (1993) Isolated congenital ACTH deficiency: a cleavage enzyme defect? Clin Endocrinol (Oxf) 39: 381–385.
- 27 Salenave S, Boyce AM, Collins MT, Chanson P (2014) Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab 99: 1955–1969.

