Abstract
The purpose of this study was to determine the efficacy and safety of tolvaptan in Japanese patients with hyponatremia secondary to syndrome of inappropriate secretion of antidiuretic hormone (SIADH). This multicenter, open-label, dose-escalation, phase III study enrolled Japanese patients (20–85 years old) with hyponatremia secondary to SIADH who were unresponsive to fluid restriction. Oral tolvaptan was administered for up to 30 days, initially at 7.5 mg/day, but escalated daily as necessary, based on the serum sodium concentration and safety, over the first 10 days until the optimal maintenance dose was determined for each patient (maximum 60 mg/day). The primary endpoint was the proportion of patients with normalized serum sodium concentration on the day after the final tolvaptan dose. Secondary endpoints included the mean change in serum sodium concentration from baseline on the day after the final dose. Sixteen patients (male, 81.3%; mean ± standard deviation age 71.9 ± 6.1 years) received tolvaptan treatment and 11 patients completed the study with one patient re-administered tolvaptan in the treatment period. Serum sodium concentrations normalized in 13 of 16 (81.3%) patients on the day after the final tolvaptan dose. The mean change in serum sodium concentration from baseline on the day after the final dose was 11.0 ± 4.3 mEq/L. Adverse events considered related to tolvaptan (10 [62.5%] patients) were generally of mild to moderate severity. Oral tolvaptan corrects hyponatremia in Japanese patients with SIADH with a similar efficacy and safety profile as that noted in non-Japanese patients.
SYNDROME OF INAPPROPRIATE SECRETION OF ANTIDIURETIC HORMONE (SIADH) is a disorder in which vasopressin is inappropriately released beyond the physiological regulatory mechanism, due to various underlying diseases [1, 2]. The causes of SIADH can be roughly classified into two types: the ectopic production of vasopressin (i.e., ectopic vasopressin-producing tumors) and excess endogenous vasopressin secretion associated with central nervous system (CNS) disorders and pulmonary diseases [1, 3].
The aim of treatment for SIADH is to correct the low sodium levels, improve the CNS-related symptoms, and prevent cerebral edema. For this purpose, elimination of the cause (e.g. treatment of the underlying disease) is the first priority. However, as the underlying disease may not be improved, or the symptoms of SIADH may be prolonged after treatment of the underlying disease, palliative therapy is the mainstay for the treatment of hyponatremia. In general, fluid restriction is the basic treatment for hyponatremia with mild to moderate reduced sodium levels, regardless of the underlying disease; however, fluid restriction commonly has a delayed onset of effect, hinders treatment of the underlying disease by infusion, and can impair the patient’s quality of life due to severe thirst [4, 5]. Although fluid restriction is identified in published guidelines as the first-line therapy for mild-to-moderate hyponatremia secondary to SIADH [5], the treatment was ineffective in more than half of cases in the USA and European Hyponatremia Registry [6].
A logical therapeutic approach for SIADH is to target vasopressin, which is the mechanism responsible for the development of hyponatremia. Vasopressin receptor antagonists target the causative mechanism of hyponatremia by competitively binding to the V2 receptor, displacing vasopressin and increasing free water clearance [1]. Tolvaptan is an oral vasopressin receptor antagonist with a high affinity for the V2 receptor and has been shown to promote aquaresis resulting in the increased excretion of free water [7, 8]. Two large, international, randomized controlled studies have demonstrated the efficacy of tolvaptan compared with placebo in patients with hyponatremia (SALT-1 and SALT-2) [7]. Based on these phase 3 studies, tolvaptan has been approved in the USA for the treatment of hyponatremia, including in patients with heart failure and SIADH, and in the European Union for the treatment of hyponatremia secondary to SIADH. However, the efficacy and safety of tolvaptan have not been demonstrated in Japanese patients. In the absence of treatments for most cases of SIADH in Japan, the Japan Endocrine Society asked the Ministry of Health, Labour and Welfare (MHLW) in 2013 to support the development of tolvaptan for hyponatremia secondary to SIADH, and the MHLW made a similar request to the tolvaptan development company (Otsuka Pharmaceutical Co., Ltd.) in 2015. As a result of this, clinical trials with tolvaptan have been conducted in Japan since 2016.
The objective of this study was to determine the efficacy and safety of tolvaptan oral tablets (7.5–60 mg/day) for up to 30 days in Japanese patients with hyponatremia secondary to SIADH.
Materials and Methods
Study design
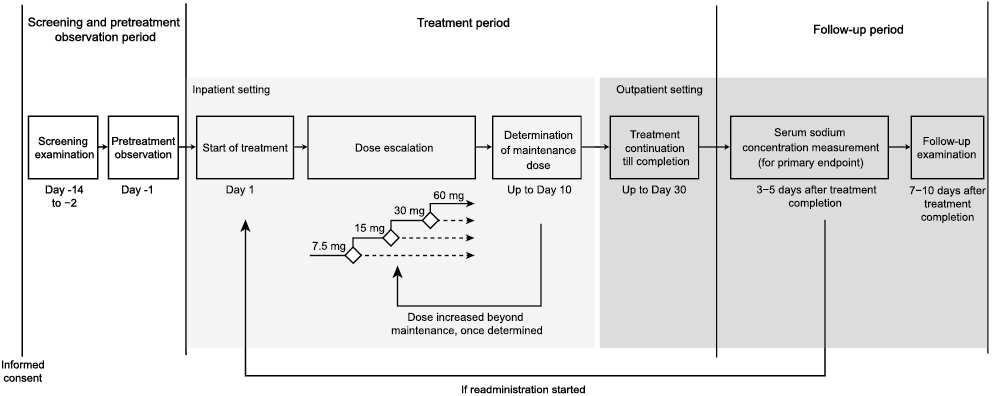
This multicenter, open-label, dose-escalation, phase 3 study enrolled Japanese patients with hyponatremia secondary to SIADH (NCT03048747). Eligible patients were either inpatients or outpatients who agreed to be hospitalized for the trial, aged 20–85 years (inclusive) at the time of informed consent with (i) a definite diagnosis of SIADH according to the criteria in the “Diagnostic and Treatment Manual of the Hypersecretion of Vasopressin (SIADH), Revised in 2011”, (ii) unresponsiveness (serum sodium concentration of <135 mEq/L) to fluid restriction (total daily fluid intake of ≤20 mL/kg body weight) for at least 7 consecutive days at the time of informed consent, (iii) fluid restriction status from the time of the screening examination until the pretreatment observation (day before tolvaptan initiation) with sustained low serum sodium concentration (<135 mEq/L at both the pretreatment observation and at predose on Day 1 of the treatment period and increased by <5 mEq/L compared with the screening examination).
Key exclusion criteria included patients with transient hyponatremia due to drugs, who do not feel thirsty or have difficulty drinking water, with urinary tract obstruction, with serum sodium levels <120 mEq/L with neurologic deficits including apathy, confusion, and seizures. A full list of exclusion criteria can be found in Supplementary Table 1.
The study was comprised of four periods: screening, pre-dose observation, treatment (maximum of 30 days, including 1 day after final dose) and follow-up (7–10 days after treatment). During the treatment phase, tolvaptan was given at 7.5 mg/day (oral tablet after breakfast) on Day 1 and increased to 15 mg/day, followed by 30 mg/day then up to a maximum dose of 60 mg/day on or after Day 2, to determine the maintenance dose in each patient (Supplementary Fig. 1). Doses were escalated if the serum sodium concentration was <130 mEq/L at Day 2 or <135 mEq/L at Day 3 and the increase in serum sodium concentration from predose on the previous day was <5 mEq/L. Once the maintenance dose had been determined (whereby the serum sodium was ≥135 mEq/L, or further escalation was inappropriate due to safety concerns or the maximum dose of tolvaptan 60 mg/day had been reached), treatment was continued at that dose throughout the treatment period. The maintenance dose could be reduced if a safety concern occurred, or increased if the patient was unresponsive to treatment due to exacerbation of underlying disease or other causes, and if such an increase did not pose a safety problem. Tolvaptan treatment was started, increased and/or resumed while the patients were in the hospital. Patients could be discharged and managed as outpatients if they met ambulatory transition criteria once the maintenance dose of tolvaptan had been determined. The ambulatory transition criteria required that the serum sodium concentration before the administration of tolvaptan did not increase by more than 2 mEq/L compared with the serum sodium concentration before the administration of tolvaptan on the previous day. To avoid very rapid and potentially dangerous increases in sodium concentration, fluid restriction was avoided for the 24-hour period following tolvaptan administration on Day 1. After Day 2 of administration, the participating investigators set an upper limit of water restriction (the upper limit being the amount of water consumed on Day 1 of administration) according to the patient’s condition, and instructed patients not to exceed this limit of water intake, including after discharge. Finally, treatment was scheduled for 30 days, but could be stopped if the patient had normalized serum sodium (≥135 mEq/L) and no subjective symptoms of hyponatremia and it was considered that the serum sodium concentration was unlikely to decrease after stopping tolvaptan. Treatment could be resumed if hyponatremia recurred within 30 days of the initial tolvaptan treatment period. Treatment was discontinued if the patient requested it, if they developed adverse events (AEs), laboratory test values for liver function (aspartate aminotransferase [AST] or alanine aminotransferase [ALT]) were ≥3 times the upper limit of normal (ULN), or when serum sodium concentration increased by ≥12 mEq/L from predose-value within 24 hours after tolvaptan administration or exceeding 145 mEq/L during the treatment period.
Concomitant use of demeclocycline, lithium carbonate, urea, intravenous saline, hypertonic saline, or drugs that had potent inhibitory or inductive effects on CYP3A4, was prohibited from the day before the start of study drug administration until the end of study drug administration.
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation (ICH 1996) Good Clinical Practice (GCP) Consolidated Guideline and applicable local laws and regulatory requirements. The protocol and the informed consent form were reviewed and approved by the institutional review board for each investigational site. Institutional Review Board (IRB) approvals were obtained from 31 hospitals.
Study variables
The primary endpoint was the proportion of patients with normalized serum sodium concentration on the day after the final administration of tolvaptan. Secondary endpoints included change in serum sodium concentration from baseline, time course of serum sodium concentration, and changes in clinical symptoms associated with hyponatremia. Serum sodium concentrations were measured regularly during the treatment period as well as 3–5 days and 7–10 days after completion of tolvaptan treatment during the follow-up period. Additional pharmacodynamic parameters were also assessed: daily urine volume, daily fluid intake and daily fluid balance.
Safety assessments included the collection of AEs and AEs related to tolvaptan, defined as AEs with a reasonable possibility of a temporal and causal relationship between the administration of tolvaptan and the AE. All AEs were recorded using system organ categories (SOC) and preferred terms (PT) defined in the Medical Dictionary for Regulatory Activities (MedDRA) version 21.1. In addition, clinical laboratory tests, body weight and vital signs, 12-lead electrocardiogram (ECG) and liver function tests were also conducted.
Statistical analysis
The efficacy analysis set consists of all patients who received ≥1 dose of tolvaptan and had post-dose serum sodium concentration data. The safety analysis set comprised all patients who received ≥1 dose of tolvaptan while the pharmacodynamic analysis set comprised those who received ≥1 dose of tolvaptan and had post-dose pharmacodynamic data. Values are given as mean ± standard deviation (SD) unless otherwise stated.
Results
Patients
Of 31 patients screened, 16 patients received tolvaptan treatment and 11 (68.8%) patients completed the study (Fig. 1). One patient (6.3%) was re-administered tolvaptan. Five patients in total discontinued the study for the reasons described below in “Safety”.
Baseline patient characteristics are shown in Table 1. Thirteen patients were male (81.3%) and 10 patients were diagnosed with SIADH linked to ectopic production of vasopressin (62.5%); the mean ± SD serum sodium concentration for the overall population was 126.9 ± 4.2 mEq/L.
Table 1
Patient characteristics at baseline (efficacy analysis set)
| Baseline characteristic |
Tolvaptan (N = 16) |
| Males, n (%) |
13 (81.3) |
| Age in years, mean ± SD |
71.9 ± 6.1 |
| Body weight in kg, mean ± SD |
54.0 ± 14.3 |
| BMI, kg/m2, mean ± SD |
21.0 ± 4.7 |
| Underlying cause, n (%) |
|
| Ectopic production of vasopressin |
10 (62.5) |
| Other |
6 (37.5) |
| Serum sodium concentration in mEq/L, mean ± SD (min, max) |
126.9 ± 4.2 (119.0, 133.0) |
| Serum sodium concentration, n (%) |
|
| <130 mEq/L |
10 (62.5) |
| ≥130 mEq/L |
6 (37.5) |
BMI, body mass index; SD, standard deviation
The individual maintenance dose of tolvaptan was 7.5 mg (n = 11) or 15 mg (n = 4), but could not be determined in one patient for whom administration of tolvaptan was discontinued on the day after the first administration. After determination of the individual maintenance dose, the dose was changed in six patients; the dose at final administration was 7.5 mg (n = 8), 15 mg (n = 6) or 60 mg (n = 1).
Efficacy
On the day after the final dose of tolvaptan, serum sodium concentrations normalized in 13 of 16 (81.3%) patients (primary efficacy endpoint) whereas, at follow-up, this proportion had decreased to four of 15 (26.7%) patients (Fig. 2). The mean change in serum sodium concentration from baseline on the day after the final dose was 11.0 ± 4.3 mEq/L. The mean serum sodium concentration increased by 6.9 mEq/L from baseline on the day after the first administration (predose on Day 2) and an increase of 8–10 mEq/L above baseline was maintained after Day 3. The time-course of mean and individual serum sodium concentrations consistently increased from baseline during the treatment period, followed by a decrease in most patients after the end of treatment (Fig. 3).

A high proportion of patients had normalized serum sodium concentrations on the day after the final tolvaptan dose across the three final doses (Supplementary Table 2). There was also a high proportion of patients with normalized serum sodium concentration on the day after the final tolvaptan administration regardless of the underlying cause leading in SIADH (ectopic production of vasopressin or other) and among those with baseline levels below and above 130 mEq/L (Supplementary Table 3). Tolvaptan treatment was associated with an increase in mean urine volume (+2,316.5 ± 1,631.6 mL/day) and fluid intake (+1,521.6 ± 1,421.4 mL/day), and a decrease in mean fluid balance (−815.1 ± 891.2 mL/day), body weight ( –1.18 ± 0.81 kg) and urine osmolality (−343.2 ± 158.6 mOsm/kg) at Day 1 (Fig. 4).

At baseline, six of 16 (37.5%) patients had clinical symptoms associated with hyponatremia, and some patients had more than one symptom. The baseline clinical symptoms were malaise (n = 6), loss of appetite (n = 4), vomiting (n = 1) and consciousness disorder (n = 1). Two patients showed improvement and the symptoms of other two were unchanged. Although worsening of malaise and loss of appetite were both observed in one patient, these events occurred in the patient who died during the treatment period due to progression of the underlying disease (lung adenocarcinoma). Symptoms could not be evaluated in one patient who was unconscious (Supplementary Table 4).
Safety
The mean duration of tolvaptan treatment was 22.6 ± 10.1 days (median 29.0 days) and 11 (68.8%) patients were given tolvaptan for between 22 and 30 days.
Overall, a total of 78 AEs were experienced by 15 (93.8%) patients (Table 2; the incidence of adverse events occurring in ≥2 patients is provided in Supplementary Table 5). Five patients experienced serious AEs, including one patient (6.3%) who experienced back pain and a lung adenocarcinoma, then died on Day 19 due to lung adenocarcinoma. The other serious AEs were transient loss of consciousness (n = 1) occurring 6 days after completing tolvaptan treatment; bacteremia, hemorrhage and paralysis of the recurrent laryngeal nerve (n = 1); supraventricular and ventricular extrasystoles (n = 1); lung adenocarcinoma and circulatory collapse (n = 1). Of the serious AEs, only the cardiac arrhythmias were considered to be related to tolvaptan and these resolved after the dose of tolvaptan was reduced.
Table 2
Summary of adverse events (AEs) in safety analysis set
|
Tolvaptan (N = 16), n (%) |
| Patients with AEs |
15 (93.8) |
| AEs, number of events |
78 |
| Patients with tolvaptan-related AEs |
10 (62.5) |
| Deaths |
1 (6.3) |
| Patients with serious AEs |
5 (31.3) |
| Patients with tolvaptan-related serious AEs |
1 (6.3) |
| Patients with severe AEs |
2 (12.5) |
| Patients with tolvaptan-related severe AEs |
0 (0.0) |
| Patients with AEs leading to discontinuation |
5 (31.3) |
| Patients with tolvaptan-related AEs leading to discontinuation |
2 (12.5) |
Five (31.3%) patients had AEs leading to treatment discontinuation (hepatic function abnormal n = 1 [6.3%], increased blood creatinine n = 1 [6.3%], lung adenocarcinoma n = 2 [12.5%] and rapid correction of hyponatremia n = 1 [6.3%]). In the patient with rapid correction of hyponatremia, the change in serum sodium concentration measured in the hospital was +12 mEq/L (117 mEq/L to 129 mEq/L) over 24 hours from initial tolvaptan administration. However, the true change (when measured at a different laboratory) was +11 mEq/L from 119 mEq/L to 130 mEq/L, and it was judged mild and non-serious. Osmotic demyelination syndrome did not occur in this patient.
Table 3 describes AEs considered to be related to tolvaptan, of which all were mild or moderate. Thirst was the most frequently reported AE, occurring in three (18.8%) patients. No significant changes were observed in vital signs and ECG parameters. One patient was treated with the highest dose (60 mg) from Day 8 to 30 of administration, but there were no safety concerns noted during the administration period.
Table 3
Incidence of adverse events (AEs) considered to be related to tolvaptan (safety analysis set)
|
Tolvaptan (N = 16), n (%) |
| Patients with any tolvaptan-related AEsa |
10 (62.5) |
| Cardiac disorders |
1 (6.3) |
| Supraventricular extrasystoles |
1 (6.3) |
| Ventricular extrasystoles |
1 (6.3) |
| Gastrointestinal disorders |
2 (12.5) |
| Constipation |
1 (6.3) |
| Dry mouth |
1 (6.3) |
| General disorders and administrative site conditions |
3 (18.8) |
| Pyrexia |
1 (6.3) |
| Thirst |
3 (18.8) |
| Investigations |
5 (31.3) |
| Alanine aminotransferase increased |
1 (6.3) |
| Blood creatinine increased |
2 (12.5) |
| Weight decreased |
2 (12.5) |
| Metabolism and Nutrition Disorders |
1 (6.3) |
| Dehydration |
1 (6.3) |
| Nervous System Disorders |
1 (6.3) |
| Headache |
1 (6.3) |
| Renal and Urinary Disorders |
1 (6.3) |
| Pollakiuria |
1 (6.3) |
| Surgical and Medical Procedures |
1 (6.3) |
| Rapid correction of hyponatremia |
1 (6.3) |
| Vascular Disorders |
1 (6.3) |
| Hypotension |
1 (6.3) |
a Patients were counted once, per term, for the most severe of multiple occurrences of a specific MedDRA preferred term.
One patient experienced serious AEs of lung adenocarcinoma and circulatory collapse on Day 5 that resulted in the discontinuation of tolvaptan. The outcome of both events was death on Day 26 (after discontinuation of tolvaptan). It was confirmed that the primary cause of death was lung adenocarcinoma in the right inferior lobe. The investigator assessed both events as severe and not related to tolvaptan. The serum sodium level on the day after the last tolvaptan administration was 134 mEq/L, but increased during the follow-up period after discontinuation (3 days after discontinuation: 141 mEq/L, 7 days after discontinuation: 153 mEq/L). This case was considered an adverse event of hypernatremia. Such an event was causally related to the serious condition of original disease of lung cancer, but was not considered to have been caused by tolvaptan treatment.
Three patients were treated with tolvaptan in combination with loop diuretics. All three of these patients (100.0%) developed adverse events compared with 12 of the 13 patients (92.3%) who did not receive concomitant loop diuretics. The same adverse event was not observed in all three patients treated with tolvaptan in combination with loop diuretics. There was no major difference in the incidence of adverse events between patients with and without loop diuretics.
Discussion
Our study showed that in Japanese patients with hyponatremia due to SIADH, oral tolvaptan (7.5–60 mg/day) for up to 30 days improved serum sodium concentrations and was well tolerated without new safety concerns. This increase in serum sodium concentration was observed immediately after the start of tolvaptan administration and continued throughout the treatment period; a high proportion of patients achieved normalized serum sodium concentrations. Moreover, the efficacy and safety observed in Japanese patients with SIADH in this study were similar to other results in similar populations in the USA, Canada and European countries (subgroup analysis of SALT studies) [7], the UK [9], China [10], and Korea [11].
The starting dose of tolvaptan for the SIADH patients in previous clinical trials was 15 mg [7, 10-12], but 7.5 mg in this study. Previously, Italian researchers have investigated the efficacy and safety of two different doses of tolvaptan (7.5 and 15 mg) in patients with hypervolemic or euvolemic hyponatremia [13]. The median increase in serum sodium was approximately two-fold higher in the group receiving tolvaptan 15 mg versus the 7.5 mg group (12 vs. 6 mEq/L, respectively; p = 0.03). These data suggest that a starting daily dose of 7.5 mg tolvaptan is both effective and safe, whereas 15 mg carries a risk of overcorrection. Similar conclusions were drawn by the authors of a retrospective study conducted in 37 patients with euvolemic hyponatremia treated with either 7.5 mg or 15 mg tolvaptan [14]. In that study, a rapid increase in serum sodium of 17 and 18 mEq/L, respectively, within the first 24 hours was seen in two of the patients who received 15 mg tolvaptan, leading these authors to recommend a starting dose of tolvaptan 7.5 mg in patients with SIADH. In the current Japanese study, using a starting dose of tolvaptan 7.5 mg showed an average increase in serum sodium of 6.9 mEq/L (range, 3–11 mEq/L) at 24 hours after administration. Furthermore, the 7.5 mg dose was the final dose for the majority of patients and was associated with good efficacy, suggesting that this dose may be the preferred starting dose for Japanese patients with hyponatremia caused by SIADH.
As noted previously, there are a number of risk factors for rapid correction of serum sodium concentration by tolvaptan, including low serum sodium (≤125 mEq/L) [11, 15], starting dose of tolvaptan (≥15 mg/day) [13, 14, 16, 17], low BUN (≤10 mg/dL) [15], as well as low body mass index and body weight [11]. In addition, patients with severe hyponatremia (e.g. ≤110 mEq/L), hypokalemia, undernutrition, alcoholism, and liver disorder are at high risk of osmotic demyelination syndrome due to rapid correction of serum sodium concentration [18, 19]. In this study, rapid correction of hyponatremia within 24 hours after tolvaptan administration was observed in one patient and was considered non-serious. At baseline, the serum sodium concentration was 119 mEq/L, which was the lowest concentration of all participants in this study. In patients at risk of rapid correction of serum sodium levels, it has been suggested that starting tolvaptan at a dose of 3.75 mg should be considered [17]. In addition, a rapid decrease in circulating plasma volume associated with rapid diuresis can lead to hemoconcentration, and this is of concern, particularly in the elderly. As asymptomatic or only mildly symptomatic chronic hyponatremia (likely due to a chronic disease progression) does not require rapid correction, more gradual correction of hyponatremia would be appropriate in this population. Therefore, starting tolvaptan at a lower dose (3.75 mg) should also be considered for this group of patients. If the desired response is not achieved with an initial tolvaptan dose of 3.75 mg, increasing the dose may be necessary, based on results of urine output and/or serum sodium levels measured before each dose, until the target increase in serum sodium level for the day is achieved.
In this study, rapid reduction in serum sodium concentrations were observed after the cessation of tolvaptan treatment. Similar changes in serum sodium concentrations have also been reported after long-term treatment with tolvaptan [20]. Thus, continued tolvaptan therapy would be necessary in order to maintain normal serum sodium concentrations; in the event of cessation of tolvaptan treatment, enriched fluid restriction should be considered.
The main strengths of this study were the multicenter, prospective nature of the study design that combined a wide dosing range and reported both clinical and pharmacodynamic outcomes. Limitations of the study include the small study sample size, absence of a comparator group and a relatively short duration of assessment of 30 days. While this study provided information on the short-term management of hyponatremia, it was not of sufficient duration to provide data on long-term treatment. Further studies in Japanese patients with SIADH, including in real-world settings, may be of benefit in providing long-term outcome data.
Conclusion
Oral tolvaptan effectively corrects hyponatremia in Japanese patients with hyponatremia due to SIADH, with approximately 80% of patients treated for up to 30 days achieving normalization in serum sodium concentration regardless of the etiology and pretreatment serum sodium concentration. Further, no significant safety concerns were identified during the trial. These efficacy and safety results reflected those noted in non-Japanese patients.
Acknowledgments
The authors thank Matt Weitz (who wrote the outline) and Mark Snape (who wrote the first draft) of Springer Healthcare Communications. This medical writing assistance was funded by Otsuka Pharmaceutical Co., Ltd.
Disclosure
Conflicts of interest/financial disclosures
The study was funded by Otsuka Pharmaceuticals Co., Ltd. H.A., K.G, S.I. are consultants for Otsuka Pharmaceutical Co., Ltd. M.T., M.M., R.W. and T.H. are employees of Otsuka Pharmaceutical Co., Ltd.
Author contributions
S.I., H.A. and K.G. provided advice regarding the study design. M.T., M.M., and R.W. designed the study, wrote the protocol, supervised the data acquisition and the monitoring activity. All authors contributed to interpretation of the data, drafting the manuscript and approved the final manuscript for submission.
Supplementary Table 1
Study exclusion criteria
| • |
Transient hyponatremia induced by drug administration. |
| • |
Dehydration. |
| • |
Hyponatremia due to heart failure, ascites associated with hepatic cirrhosis, or nephrotic syndrome. |
| • |
SIADH due to unknown etiology. |
| • |
Substantial sodium transudation due to renal sodium loss, diarrhea, or vomiting. |
| • |
Inability to sense thirst or difficulty with fluid intake. |
| • |
Impaired consciousness (coma or stupor). |
| • |
AST or ALT exceeding 3 times the upper limit of normal at the screening examination. |
| • |
History of myocardial infarction within 30 days prior to informed consent. |
| • |
History of persistent ventricular tachycardia or ventricular fibrillation despite having an implantable cardioverter defibrillator within 30 days prior to informed consent. |
| • |
Decubitus angina, exertional angina induced by even mild exertion, or severe angina such as unstable angina. |
| • |
History of cerebrovascular disorder within 30 days prior to informed consent. |
| • |
Severe psychiatric disorder. |
| • |
Systolic blood pressure of <90 mmHg at the time of the screening examination or at the pretreatment observation on the day before start of tolvaptan administration |
| • |
History of hypersensitivity or idiosyncratic reaction to benzazepine or benzazepine derivatives such as benazepril. |
| • |
History of drug abuse within 1 year prior to informed consent or with current alcohol dependence. |
| • |
Poorly controlled diabetes mellitus, defined as fasting blood glucose level of >300 mg/dL. |
| • |
Urinary tract obstruction. |
| • |
Participated in any other clinical trial within 30 days prior to informed consent. |
| • |
Participated in another clinical trial of tolvaptan or who have previously received tolvaptan. |
| • |
Terminal stage of the disease or in a moribund state with an expected survival of less than 3 months as judged by the investigator or subinvestigator. |
| • |
Serum sodium concentration of <120 mEq/L associated with neurologic impairment, including apathy, confusion, or seizures. |
| • |
Progressive or episodic neurological disorder, including multiple sclerosis or with a history of multiple strokes. |
| • |
Acute or transient hyponatremia due to head trauma or postoperative condition. |
| • |
Receiving intravenous infusion fluid at a KVO volume (at the minimum fluid volume for preventing blood clots after the planned volume of fluid infusion) or higher. |
| • |
Hyponatremia due to an artefact in laboratory tests (eg, a high blood glucose level of >300 mg/dL). |
| • |
Receiving AVP or an AVP analog for the treatment of the disease. |
| • |
Receiving other drugs for the treatment of hyponatremia, including demeclocycline, lithium carbonate, or urea within 7 days prior to start of tolvaptan administration. |
| • |
Concomitant use of hypertonic saline for correction of serum sodium concentration within 7 days prior to informed consent or who were likely to require intravenous hypertonic saline infusion for correction of symptomatic or asymptomatic severe hyponatremia during the trial period. |
| • |
Severe pulmonary hypertension: patients whose disease state could have been expected to worsen due to sudden changes in body fluid volume and blood pressure. |
| • |
Breast-feeding at the planned start of tolvaptan administration or who were judged by the investigator or subinvestigator to be possibly pregnant based on the results of the pregnancy test at the screening examination. |
| • |
Male patients or female patients of childbearing potential who were sexually active and did not agree to practice birth control as specified or to remain abstinent during the trial and until 30 days after final tolvaptan administration. |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; AVP, arginine vasopressin; KVO, keep vein open; SIADH, syndrome of inappropriate secretion of antidiuretic hormone
Supplementary Table 2
Proportion of patients with normalized serum sodium concentrations on the day after final tolvaptan dose
| Final dose |
Na |
nb |
% |
CI (lower, upper) |
| 7.5 mg |
8 |
8 |
100.0 |
63.1, 100.0 |
| 15 mg |
6 |
4 |
66.7 |
22.3, 95.7 |
| 60 mg |
1 |
1 |
100.0 |
2.5, 100.0 |
CI, 95% confidence interval.
Na = total number of treated patients with serum sodium concentration at predose on Day 1 of the treatment period of <135 mEq/L.
nb = the number of patients with normalized serum sodium concentration of ≥135 mEq/L.
Supplementary Table 3
Proportion of patients with normalized serum sodium concentrations by underlying cause of SIADH and baseline serum sodium concentration, the day after final tolvaptan dose (efficacy analysis set)
| Parameter |
Na |
nb |
% |
CI (lower, upper) |
| Underlying cause |
|
|
|
|
| Ectopic production of vasopressin |
10 |
9 |
90.0 |
55.5, 99.7 |
| Other |
6 |
4 |
66.7 |
22.3, 95.7 |
| Baseline serum sodium |
|
|
|
|
| <130 mEq/L |
10 |
7 |
70.0 |
34.8, 93.3 |
| ≥130 mEq/L |
6 |
6 |
100.0 |
54.1, 100.0 |
Na = total number of treated patients with serum sodium concentration at predose on Day 1 of the treatment period <135 mEq/L.
nb = the number of patients with normalized serum sodium concentration as ≥135 mEq/L.
Supplementary Table 4
Clinical symptoms throughout the study period among the 6 patients who had symptoms at baseline
|
|
Clinical symptoms |
| Patient |
Underlying cause of SIADH |
Baseline |
Day after final dose |
| 1 |
Ectopic production
of vasopressin |
Loss of appetite, g1
Vomiting, g1
Malaise, g1 |
Loss of appetite, nothing
Vomiting, nothing
Malaise, g1 |
| 2 |
Other |
Malaise, g1 |
Malaise, nothing |
| 3 |
Ectopic production
of vasopressin |
Loss of appetite, g1
Malaise, g1 |
Loss of appetite, g1
Malaise, g1 |
| 4 |
Other |
Mild consciousness disorder
Malaise, g1 |
Mild consciousness disorder
Malaise, g1 |
| 5 |
Other |
Loss of appetite, g3
Malaise, g1 |
N.E. |
| 6 |
Ectopic production of vasopressin |
Loss of appetite, g1
Malaise, g1 |
Loss of appetite, g2
Malaise, g2 |
g1, grade 1 (mild); g2, grade 2 (moderate); g3, grade 3 (severe); N.E., not evaluated due to severe consciousness disorder.
Supplementary Table 5
Incidence of adverse events occurring in ≥2 patients (safety analysis set)
|
Tolvaptan (N = 16) n (%) |
| Blood and lymphatic system disorders |
|
| Neutropenia |
2 (12.5) |
| Gastrointestinal disorders |
|
| Constipation |
2 (12.5) |
| Nausea |
2 (12.5) |
| Vomiting |
2 (12.5) |
| General disorders and administration site conditions |
|
| Pyrexia |
2 (12.5) |
| Thirst |
3 (18.8) |
| Infections and infestations |
|
| Gingivitis |
2 (12.5) |
| Injury, poisoning, and procedural complications |
|
| Fall |
2 (12.5) |
| Investigations |
|
| Blood creatinine increased |
2 (12.5) |
| Platelet count decreased |
2 (12.5) |
| Weight decreased |
2 (12.5) |
| Metabolism and nutrition disorders |
|
| Decreased appetite |
3 (18.8) |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) |
|
| Lung adenocarcinoma |
2 (12.5) |
| Respiratory, thoracic, and mediastinal disorders |
|
| Hiccups |
2 (12.5) |
References
- 1 Cuesta M, Garrahy A, Thompson CJ (2016) SIAD: practical recommendations for diagnosis and management. J Endocrinol Invest 39: 991–1001.
- 2 Peri A, Giuliani C (2014) Management of euvolemic hyponatremia attributed to SIADH in the hospital setting. Minerva Endocrinol 39: 33–41.
- 3 Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH (2007) Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med 120: S1–S21.
- 4 Ishikawa S (2011) SIADH. In: Orita Y (ed) Vasopressin and its receptor antagonism basic principles and clinical applications. Osaka: Medical Review, 141–149 (In Japanese).
- 5 Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, et al. (2014) Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J Endocrinol 170: G1–G47.
- 6 Verbalis JG, Greenberg A, Burst V, Haymann JP, Johannsson G, et al. (2016) Diagnosing and treating the syndrome of inappropriate antidiuretic hormone secretion. Am J Med 129: 537.e9–537.e23.
- 7 Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, et al. (2006) Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 355: 2099–2112.
- 8 Yamamura Y, Nakamura S, Itoh S, Hirano T, Onogawa T, et al. (1998) OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther 287: 860–867.
- 9 Humayun MA, Cranston IC (2017) In-patient tolvaptan use in SIADH: care audit, therapy observation and outcome analysis. BMC Endocr Disord 17: 69.
- 10 Chen S, Zhao JJ, Tong NW, Guo XH, Qiu MC, et al. (2014) Randomized, double blinded, placebo-controlled trial to evaluate the efficacy and safety of tolvaptan in Chinese patients with hyponatremia caused by SIADH. J Clin Pharmacol 54: 1362–1367.
- 11 Han SW, Yi JH, Kang KP, Kim HY, Kim SW, et al. (2018) Safety and efficacy of tolvaptan in Korean patients with hyponatremia caused by the syndrome of inappropriate antidiuretic hormone. J Korean Med Sci 33: e112.
- 12 Verbalis JG, Adler S, Schrier RW, Berl T, Zhao Q, et al. (2011) Efficacy and safety of oral tolvaptan therapy in patients with the syndrome of inappropriate antidiuretic hormone secretion. Eur J Endocrinol 164: 725–732.
- 13 Castello LM, Baldrighi M, Panizza A, Bartoli E, Avanzi GC (2017) Efficacy and safety of two different tolvaptan doses in the treatment of hyponatremia in the emergency department. Intern Emerg Med 12: 993–1001.
- 14 Harbeck B, Lindner U, Haas CS (2016) Low-dose tolvaptan for the treatment of hyponatremia in the syndrome of inappropriate ADH secretion (SIADH). Endocrine 53: 872–873.
- 15 Morris JH, Bohm NM, Nemecek BD, Crawford R, Kelley D, et al. (2018) Rapidity of correction of hyponatremia due to syndrome of inappropriate secretion of antidiuretic hormone following tolvaptan. Am J Kidney Dis 71: 772–782.
- 16 Tzoulis P, Waung JA, Bagkeris E, Carr H, Khoo B, et al. (2016) Real-life experience of tolvaptan use in the treatment of severe hyponatraemia due to syndrome of inappropriate antidiuretic hormone secretion. Clin Endocrinol (Oxf) 84: 620–626.
- 17 Sterns RH (2018) Tolvaptan for the syndrome of inappropriate secretion of antidiuretic hormone: is the dose too high? Am J Kidney Dis 71: 763–765.
- 18 Research on Hypothalamic Pituitary Dysfunction Study Group (2019) Guide to Diagnosis and Treatment of Hypothalamic Pituitary Dysfunction (2018 updated). Folia Endocrinologica Japonica 95: 1–60 (In Japanese).
- 19 Seay NW, Lehrich RW, Greenberg A (2020) Diagnosis and management of disorders of body tonicity—hyponatremia and hypernatremia: core curriculum 2020. Am J Kidney Dis 75: 272–286.
- 20 Berl T, Quittnat-Pelletier F, Verbalis JG, Schrier RW, Bichet DG, et al. (2010) Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol 21: 705–712.