Abstract
Chronic pancreatitis (CP) is a chronic inflammatory and fibrotic disease of the pancreas, and activated pancreatic stellate cells (PSCs) play a vital role in the progression of pancreatic fibrosis in CP. It has been reported that long non-coding RNA small nucleolar RNA host gene 11 (SNHG11) is highly expressed in chronic pancreatitis (CP) patients. However, the role of SNHG11 in CP progression is unclear. The purport of the study was to survey the role of SNHG11 in CP. We employed transforming growth factor (TGF)-beta1 (TGF-β1) to activate human pancreatic stellate cells (PSCs). Expression of SNHG11 was assessed with qRT-PCR. Loss-of-function experiments were executed to evaluate the effects of SNHG11 on the proliferation and migration of TGF-β1-treated PSCs. Some protein levels were detected by western blotting. The regulatory mechanism of SNHG11 was verified by the dual-luciferase reporter and RIP assays. As a result, SNHG11 was upregulated in plasma of CP patients and TGF-β1-treated PSCs. Also, SNHG11 inhibition reduced TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs. Mechanistically, SNHG11 regulated leukemia inhibitory factor (LIF) expression by sponging miR-34b. Furthermore, miR-34b inhibitor abolished SNHG11 silencing-mediated effects on TGF-β1-treated PSC proliferation, migration, and ECM accumulation. LIF overexpression counteracted the repressive influence of miR-34b mimic on proliferation, migration, and ECM accumulation of TGF-β1-treated PSCs. In conclusion, SNHG11 knockdown reduced TGF-β1-induced PSC proliferation, migration, and ECM accumulation by the miR-34b/LIF axis.
CHRONIC PANCREATITIS (CP) is a progressive disease characterized by inflammation of the pancreas, which causes fibrosis and destroys exocrine and endocrine tissues [1]. Pancreatic fibrosis is the main risk factor for pancreatic cancer [2], which is caused by the proliferation of fibroblasts and the deposition of extracellular matrix (ECM), containing collagen fibers [3]. Pancreatic stellate cells (PSCs) are essential for maintaining normal pancreas structure [4], and activated PSCs play a key role in the development of pancreatic fibrosis [5, 6]. Therefore, it is of great significance to explore the mechanisms related to pancreatic fibrosis in CP.
Long non-coding RNAs (lncRNAs) are a type of transcripts that are longer than 200 nucleotides and do not have protein-coding capability [7]. According to the competing endogenous RNA (ceRNA) hypothesis, lncRNAs can adsorb microRNA (miRNA) by miRNA response elements (MREs), thus affect miRNA affinity with mRNA [8]. It has been proved that lncRNAs participate in diverse biological functions and disease processes [9] and act as potential biomarkers for disease diagnosis [10, 11]. Moreover, lncRNAs were revealed to be connected with pancreatic disease [12-14]. For instance, lncRNA MIAT regulated the fibrosis process of CP by modulating the activation of PSCs [15]. Small nucleolar RNA host gene 11 (SNHG11), also termed as NCRNA00101, is an lncRNA. SNHG11 has been uncovered as a latent biomarker for colorectal cancer early diagnosis [16]. Moreover, SNHG11 can contribute to the progression of hepatocellular carcinoma [17] and lung cancer [18]. However, the biological function of SNHG11 in CP was unclear.
In the current study, we explored the biological role of SNHG11 in CP. Our results suggested that SNHG11 inhibition decreased TGF-β1-induced proliferation, migration, and ECM accumulation through the miR-34b/LIF pathway in PSCs.
Materials and Methods
CP specimens
The research was authorized and supervised by the Ethics Committee of The First Affiliated Hospital of Bengbu Medical College of the Joint Service Support Force. 60 clinical plasma samples were obtained from The First Affiliated Hospital of Bengbu Medical College of which 30 were from CP patients and 30 were from healthy controls. The collected venous blood samples were placed in tubes containing ethylene diamine tetraacetic acid. Next, plasma was obtained by centrifugation at 1,600 × g for 10 min. Written informed consents were obtained from all participants.
Cell culture, treatment, and transfection
Human pancreatic stellate cells (PSCs) were bought form Procell (Wuhan, China) and cultured in DMEM/F-12 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (fetal bovine serum) (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Sigma, St. Louis, MO, USA) in a humidified atmosphere with 5% CO2 at 37°C. For transforming growth factor (TGF)-beta1 (TGF-β1) treatment, PSCs were stimulated with TGF-β1 (10 ng/mL, Sigma) for 48 h.
Small interference RNA targeting SNHG11 (si-SNHG11) and corresponding negative control (si-NC), miR-34b mimic and inhibitor (miR-34b and anti-miR-34b), and their matching negative controls (miR-NC and anti-NC) were obtained from GenePharma (Shanghai, China). Cell transfection was performed using Lipofectamine 3000 reagent (Life Technologies, Carlsbad, CA, USA). For the pcDNA-LIF (LIF) (Accession: NM_002309) plasmid, the sequence of LIF was synthesized by GenePharma and then cloned into the pcDNA3.1 vector (Thermo Fisher Scientific) using the EcoRI and XhoI restriction sites.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The RNAiso Plus (Takara, Shiga, Japan) was applied to extract total RNA. A High-Capacity complementary DNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) or miRNA First-Strand Synthesis Kit (Takara) was utilized to carry out the reverse transcription. qPCR was carried out with the SYBR Premix Ex TaqTM II kit (Takara) in the CFX96 Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA). The 2–ΔΔCt method was utilized to figure the levels of SNHG11, miR-34b, and LIF mRNA, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or U6 small nuclear RNA (snRNA) was used as an internal control. The primer sequences used in the research were displayed as below: SNHG11 (F: 5'-TGGGAGTTGTCATGTTGGGA-3', R: 5'-ACTCGTCACTCTTGGTCTGT-3'), miR-34b (F: 5'-CGCGTAGGCAGTGTCATTAGC-3', R: 5'-AGTGCAGGGTCCGAGGTATT-3'), LIF (F: 5'-AGATCAGGAGCCAACTGGCACA-3', R: 5'-GCCACATAGCTTGTCCAGGTTG-3'), GAPDH (F: 5'-GAAGGTGAAGGTCGGAGTC-3', R: 5'-GAAGATGGTGATGGGATTTC-3'), and U6 snRNA (F: 5'-GCTTCGGCAGCACATATACTAAAAT-3', R: 5'-CGCTTCACGAATTTGCGTGTCAT-3').
Cell counting kit-8 (CCK-8) assay
Transfected PSCs (5 × 103 cells/well) were seeded in 96-well plates for 24 h, 48 h, or 72 h. Then, CCK-8 solution (Beyotime, Jiangsu, China) was added to each well and kept for 4 h. The microplate reader (Bio-Rad) was applied to analyze the absorbance at 450 nm.
Wound healing assay
The transfected PSCs (2 × 105 cells/mL) were seeded in 6-well plates. When grown to 90% confluence, the cell monolayers were scraped with a sterile micropipette tip. The wound gaps were imaged with a light microscope (Olympus, Tokyo, Japan) at 0 h and 48 h.
Transwell assay
Cell migration ability was assessed with the transwell chamber (Costar, Cambridge, MA, USA). In short, 200 μL cell medium containing PSCs (1 × 105 cells) was added to upper chambers. The bottom chamber was added with cell medium (600 μL) containing FBS (10%). After culture for 24 h, the migrating cells were fixed with paraformaldehyde (5%), followed by staining with crystal violet (0.5%, Sigma). The number of migrated cells was determined by an inverted microscope (Olympus) under 100× magnification.
Western blotting
Total protein was extracted with the RIPA lysis buffer (Beyotime). After quantification with the BCA Assay Kit (Pierce, Rockford, IL, USA), the extracted total protein was separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10%, SDS-PAGE). Afterward, the separated proteins were transferred to the polyvinylidene difluoride (PVDF, Millipore, Billerica, MA, USA) membranes and then blocked with Tris Buffered Saline Tween (TBST) buffer with 5% skims milk. Next, the PVDF membranes were incubated with primary antibodies at 4°C overnight, including anti-COL1A1 (ab90395, 1:500, Abcam, Cambridge, MA, USA), alpha-smooth muscle actin (α-SMA) (ab32575, 1:1,000, Abcam), LIF (ab235908, 1:1,000, Abcam), and anti-GAPDH (ab8245, 1:500, Abcam). Next, the PVDF membranes were incubated with goat anti-mouse immunoglobulin G (IgG) (ab6708, 1:3,000, Abcam) or anti-rabbit IgG (ab97051, 1:2,000, Abcam). Finally, the immunoblot was visualized through enhanced chemiluminescence solution (Beyotime).
Nuclear-cytoplasmic fractionation assay
The PARIS kit (Life Technologies) was utilized for nuclear and cytoplasmic RNAs extraction. The levels of SNHG11 in the nuclear and cytoplasmic RNAs were analyzed via qRT-PCR, and U6 snRNA or 18S ribosomal RNA (rRNA) was used as a control for nucleus and cytoplasm, respectively. The primer sequences of 18S rRNA were F: 5'-GTGGTGTTGAGGAAAGCAGACA-3' and R: 5'-TGATCACACGTTCCACCTCATC-3'.
Dual-luciferase reporter assay
The targets of SNHG11 were predicted by miRcode and GSE25820 databases. The targets of miR-34b were predicted using starbase and GSE91035 databases. The sequences of wild type SNHG11 (SNHG11-wt), mutant SNHG11 (SNHG11-mut), LIP 3'UTR (Untranslated Region)-wt, and LIP 3'UTR-mut were synthesized and inserted into the psiCHECK-2 plasmids (Promega, Madison, WI, USA), respectively. PSCs were transfected a luciferase reporter and miR-34b mimic and miR-NC with Lipofectamine 3000. The luciferase activities were assessed with the dual-luciferase reporter assay system (Promega).
RNA pull-down assay with biotinylated SNHG11 probe
The biotinylated SNHG11 probe and Oligo probe were synthesized by Sangon Biotech (Shanghai, China). In brief, the lysates of PSCs were incubated with probe-coated beads (Sigma), which were obtaining by co-incubating biotinylated SNHG11 probe or Oligo probe with streptavidin magnetic beads (Thermo Fisher Scientific). The enrichment of miR-34b, miR-184, and miR-373 was analyzed by qRT-PCR.
RNA pull-down assay with biotinylated miR-34b probe
PSCs were transfected with biotinylated miR-34b probe (Bio-miR-346) or Oligo probe (Sangon Biotech). 48 h later, the lysates of PSCs were incubated M-280 streptavidin magnetic beads (Sigma), which were blocked with yeast tRNA (Sigma). The abundances of PALLD, LIF, VEGFA, GJC1, CALD1, MRVI1, CST1, RGS5 were analyzed by qRT-PCR.
RNA immunoprecipitation (RIP) assay
The specific binding between SNHG11 or LIP and miR-34b was verified by RIP assay with the EZ-Magna RIP Kit (Millipore). The lysate of PSCs was incubated with protein A/G sepharose beads conjugated with anti-IgG or anti-Argonaute2 (Ago2) antibodies (Sigma). The enrichment of SNHG11, LIP, or miR-34b was analyzed by qRT-PCR.
Statistical analysis
All experiments were repeated at least 3 times, and each experiment was performed in triplicate. Data were expressed as mean ± standard deviation. SPSS 20.0 software (IBM Corporation, Armonk, NY, USA) was utilized to carry out the statistical analysis. Independent Student’s t test was utilized to evaluate the differences between two groups. Analysis of variance with Tukey’s multiple comparisons test was employed to compare the difference among multiple groups. Differences were deemed significant if p < 0.05.
Results
SNHG11 knockdown reduced TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs
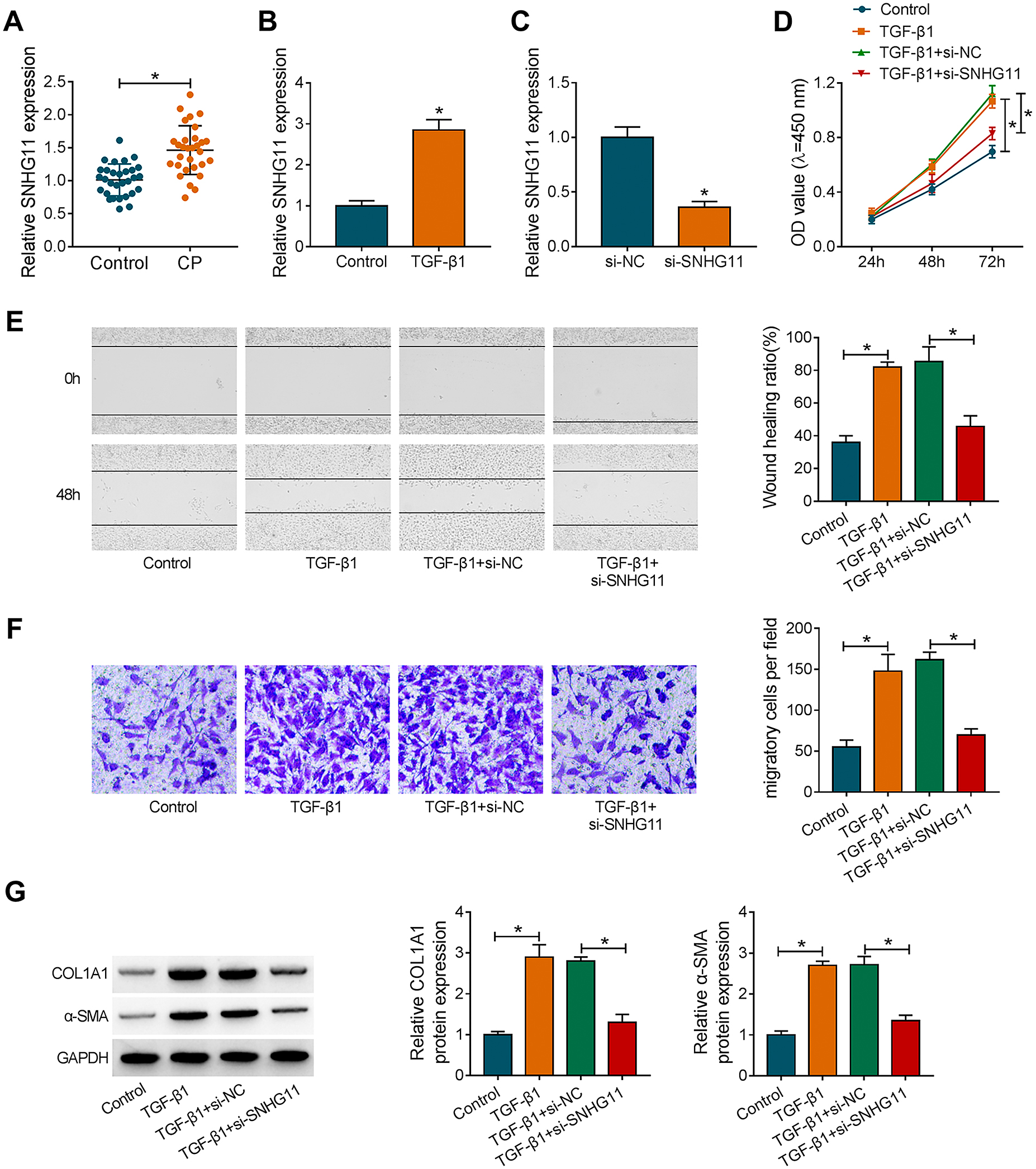
To verify the expression pattern of SNHG11 in CP, we detected SNHG11 expression in plasma of 30 CP patients and 30 healthy controls. qRT-PCR exhibited that SNHG11 was highly expressed in patients with CP relative to healthy controls (Fig. 1A). Moreover, SNHG11 expression was higher in TGF-β1-treated PSCs in contrast to the control group (Fig. 1B). To validate the function of SNHG11 in CP, we performed loss-of-function experiments. The knockdown efficiency of si-SNHG11 in PSCs was validated by qRT-PCR (Fig. 1C). As exhibited in Fig. 1D–1F, the silence of SNHG11 reduced TGF-β1-induced proliferation and migration in PSCs. Also, the upregulation of COL1A1 and α-SMA in PSCs caused by TGF-β1 stimulation was overturned after SNHG11 knockdown (Fig. 1G). Together, these results indicated that SNHG11 might be related to the fibrosis process of CP.
SNHG11 acted as a decoy for miR-34b in PSCs
To explore the regulation mechanism of SNHG11 in CP, we first analyzed the distribution of SNHG11 in PSCs. nuclear-cytoplasmic fractionation experiments manifested that SNHG11 was mainly distributed in the cytoplasm of PSCs (Fig. 2A). Through analyzing miRcode and GSE25820 databases, we found that SNHG11 might be a decoy for 3 miRNAs (miR-34b, miR-184, and miR-373), which were among the top 100 miRNAs with the most significant downregulation in CP (Fig. 2B). Moreover, miR-34b and miR-184 were markedly pulled-down by SNHG11 probe compared to the Oligo probe, particularly miR-34b (Fig. 2C). There was an apparent downregulation of miR-34b in CP patients in the GSE25820 database (Fig. 2D). The putative binding sites of SNHG11 in miR-34b and a mutant sequence were exhibited in Fig. 2E. The overexpression efficiency of miR-34b mimic in PSCs was showed in Fig. 2F. Dual-luciferase reporter assay revealed that miR-34b overexpression could reduce the luciferase intensity of the SNHG11-wt reporter in PSCs, while there was no overt difference in the SNHG11-mut reporter (Fig. 2G). RIP assay exhibited that miR-34b and SNHG11 could be co-enriched in the anti-Ago2 group in comparison to the control IgG (Fig. 2H). Also, miR-34b expression was apparently decreased in TGF-β1-treated PSCs and the plasma of CP patients (Fig. 2I and 2J). Pearson’s correlation analysis presented that there was a negative correlation between miR-34b and SNHG11 expression in plasma of CP patients (Fig. 2K). Collectively, these data indicated that SNHG11 was a sponge of miR-34b in PSCs.

Subsequently, we explored whether SNHG11 regulated TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs by adsorbing miR-34b. QRT-PCR confirmed the knockdown efficiency of anti-miR-34b in PSCs, as exhibited in Fig. 3A. We observed that the introduction of anti-miR-34b reversed the repressive effect of SNHG11 knockdown on proliferation and migration of TGF-β1-treated PSCs (Fig. 3B–3D). Also, miR-34b inhibitor overturned the downregulation of COL1A1 and α-SMA in SNHG11-silenced PSCs under TGF-β1 stimulation (Fig. 3E). These findings revealed that SNHG11 adsorbed miR-34b to regulate TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs.
MiR-34b directly targeted LIF in PSCs
To survey the downstream target of miR-34b in CP, we predicted the targets of miR-34b by starbase and GSE91035 databases. As displayed in Fig. 4A, 8 genes (PALLD, LIF, VEGFA, GJC1, CALD1, MRVI1, CST1, RGS5), which among the top 100 genes with the most significant upregulation in CP, might be targets of miR-34b. Furthermore, only 2 genes (LIF and VEGFA) could be pulled down by the miR-34b probe in the 8 predicted genes (Fig. 4B). The upregulation of LIF in CP patients in the GSE91035 database was showed in Fig. 4C. The predicted binding sequence between miR-34b and LIF 3'UTR were presented in Fig. 4D. Moreover, the luciferase activity of the LIF 3'UTR-wt reporter was reduced in miR-34b-overexpressed PSCs, but the luciferase activity of the LIF 3'UTR-mut reporter did not change (Fig. 4E). Also, miR-34b and LIF were prominently enriched in the Ago2 antibody group (Fig. 4F). As expected, miR-34b mimic repressed the levels of LIF mRNA and protein in PSCs, but miR-34b inhibitor had a reverse influence (Fig. 4G and 4H). In addition, the mRNA and protein levels of LIF were upregulated in TGF-β1-treated PSCs and the plasma of CP patients (Fig. 4I–4L). The expression of LIF mRNA was negatively correlated with miR-34b in the plasma of CP patients (Fig. 4M). These results suggested that LIF severed as a target for miR-34b in PSCs.

Based on the above findings, we further explored whether LIF was involved in miR-34b mimic-mediated PSC proliferation, migration, and ECM accumulation under TGF-β1 stimulation. After LIF transfection, the levels of LIF mRNA and protein were upregulated in PSCs (Fig. 5A and 5B). As expected, the forcing expression of LIF reversed the inhibitory effect of miR-34b mimic on proliferation and migration of TGF-β1-treated PSCs (Fig. 5C–5E). Moreover, miR-34b mimic decreased the protein levels of COL1A1 and α-SMA in TGF-β1-treated PSCs, but this decrease was restored after LIF overexpression (Fig. 5F). Together, these results indicated that miR-34b regulated TGF-β1-induced proliferation, migration, and ECM accumulation by targeting LIF in PSCs.
SNHG11 adsorbed miR-34b to regulate LIF expression in PSCs
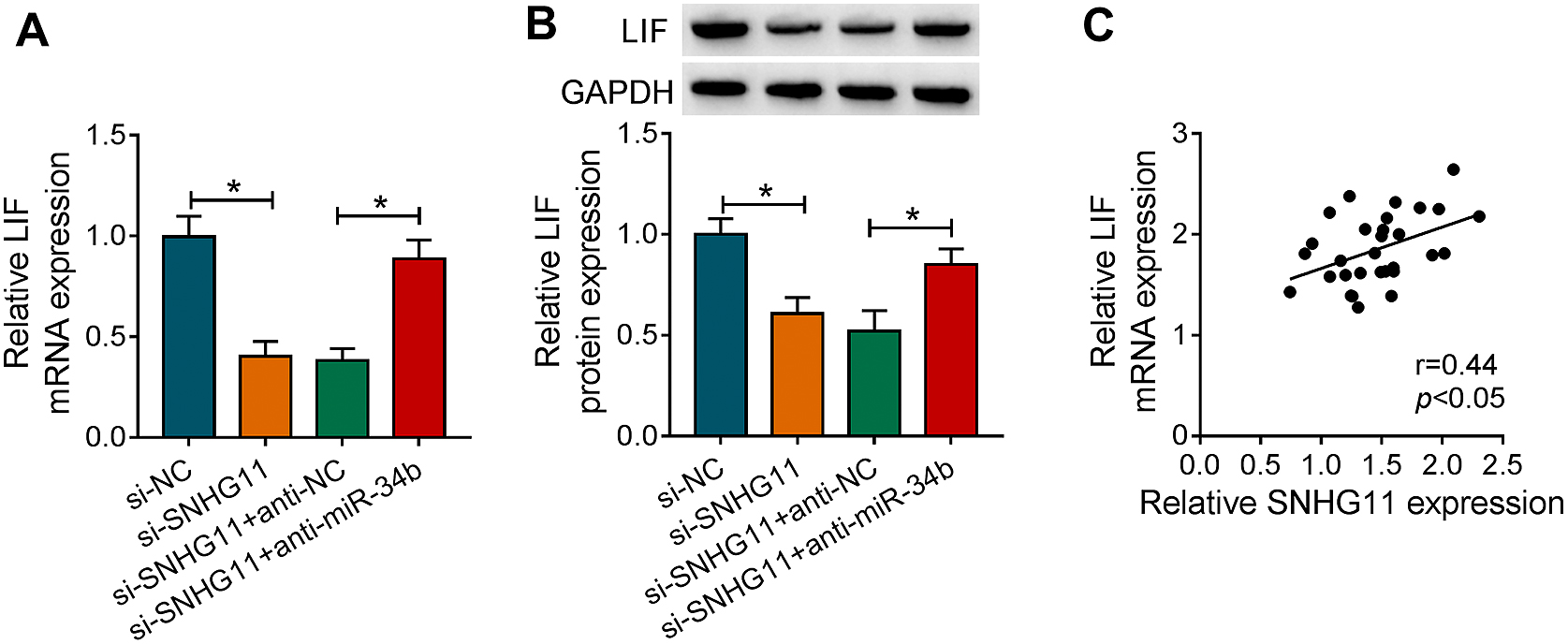
Given that SNHG11 acted as a decoy for miR-34b, which targeted LIF in PSCs. We further investigated whether SNHG11 functioned as a ceRNA to regulate LIF expression by sponging miR-34b. The results exhibited that the introduction of anti-miR-34b overturned the downregulation of LIF in si-SNHG11-transfected PSCs (Fig. 6A and 6B). Also, there was a positive correlation between LIF mRNA and SNHG11 in the plasma of CP patients (Fig. 6C). These results manifested that SNHG11 could regulate LIF expression by sponging miR-34b.
Discussion
TGF-β1 is a member of TGF-beta superfamily, which plays a role in several biological processes [19]. It has been uncovered that TGF-β1 is a key regulator of PSC activation and ECM production [20]. A previous study revealed that TGF-β1 overexpression induced CP in murine pancreas [21]. Also, inhibition of the activation of TGF-β1 precursor reduced the severity of pancreatic fibrosis [22]. α-SMA is commonly used as a marker of fibrotic activity of activated tissue fibrotic cells [23]. COL1A1, a component of ECM, is usually upregulated during the process of fibrosis [24, 25]. Herein, SNHG11 was upregulated in plasma of CP patients and TGF-β1-treated PSCs. Moreover, SNHG11 inhibition reduced proliferation, migration, and protein levels of α-SMA and COL1A1 in TGF-β1-induced PSCs. Report of Liu et al. also pointed out that SNHG11 expression was enhanced in CP patients, and SNHG11 might be a biomarker for CP diagnosis [26]. Therefore, we concluded that TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs partly depended on the mechanism regulated by SNHG11 in CP.
Increasing evidence revealed that lncRNAs could act as ceRNAs to regulate gene expression by adsorbing miRNAs in various diseases [27]. In this study, SNHG11 was mainly distributed in the cytoplasm of PSCs, implying that SNHG11 might function as a ceRNA. Though bioinformatics analysis, dual-luciferase reporter, and RIP assays, we verified SNHG11 as a decoy for miR-34b in PSCs. MiR-34b had been reported to play a tumor-suppressive role in a series cancers [28-30]. Liu et al. revealed that miR-34b severed as a tumor metastasis suppressor in pancreatic cancer through targeting Smad3 [31]. Circular RNA circ-BFAR sponged miR-34b to increase MET expression, resulting in accelerating the progression of pancreatic ductal adenocarcinoma [32]. Also, Munding et al. revered that miR-34b was downregulated in CP patients (GSE25820) [33]. In this study, we verified that miR-34b was reduced in the plasma of CP patients and TGF-β1-treated PSCs. Furthermore, miR-34b inhibitor revered SNHG11 inhibition-mediated influence on TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs. These results indicated that miR-34b might play a protective role during fibrosis progression in CP, and SNHG11 modulated TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs by sponging miR-34b.
In addition, we found that LIF acted as a target for miR-34b. LIF, a member of the interleukin-6 cytokine superfamily, has a wide range of biological activities that can affect bone metabolism, cachexia, neurodevelopment, embryogenesis, and inflammation [34]. Also, LIF derived from activated PSCs was related to the pathogenesis of pancreatic ductal adenocarcinoma [35] and LIF might be a potential therapeutic target for pancreatic ductal adenocarcinoma [36, 37]. Herein, LIF overexpression restored the suppressive impact of miR-34b mimic on TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs, indicating that miR-34b targeted LIF to modulate TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs. Notably, SNHG11 acted a ceRNA and regulated LIF expressed by sponging miR-34b in PSCs. Therefore, we concluded that SNHG11 regulated TGF-β1-induced proliferation, migration, and ECM accumulation in PSCs by the miR-34b/LIF pathway.
In conclusion, TGF-β1-induced SNHG11 sponged miR-34b to elevate LIF expression, thereby facilitating proliferation, migration, and ECM accumulation of PSCs. The research offered a new underlying mechanism by which SNHG11 regulated the fibrosis process of CP.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflict of Interest
None declared.
Acknowledgement
Thanks for all participants involved in this study.
References
- 1 Jagannath S, Garg PK (2017) Novel and experimental therapies in chronic pancreatitis. Dig Dis Sci 62: 1751–1761.
- 2 Thomas D, Radhakrishnan P (2019) Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer 18: 14.
- 3 Wang H, Jiang Y, Lu M, Sun B, Qiao X, et al. (2017) STX12 lncRNA/miR-148a/SMAD5 participate in the regulation of pancreatic stellate cell activation through a mechanism involving competing endogenous RNA. Pancreatology 17: 237–246.
- 4 Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, et al. (2017) Pancreatic stellate cell: pandora’s box for pancreatic disease biology. World J Gastroenterol 23: 382–405.
- 5 Lin Z, Zheng LC, Zhang HJ, Tsang SW, Bian ZX (2015) Anti-fibrotic effects of phenolic compounds on pancreatic stellate cells. BMC Complement Altern Med 15: 259.
- 6 Xu XF, Liu F, Xin JQ, Fan JW, Wu N, et al. (2018) Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-β1 (TGF-β1). Biochem Biophys Res Commun 501: 365–373.
- 7 Ma L, Bajic VB, Zhang Z (2013) On the classification of long non-coding RNAs. RNA Biol 10: 925–933.
- 8 Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146: 353–358.
- 9 Xu J, Bai J, Zhang X, Lv Y, Gong Y, et al. (2017) A comprehensive overview of lncRNA annotation resources. Brief Bioinform 18: 236–249.
- 10 Li L, Wang L, Li H, Han X, Chen S, et al. (2018) Characterization of LncRNA expression profile and identification of novel LncRNA biomarkers to diagnose coronary artery disease. Atherosclerosis 275: 359–367.
- 11 Chao Y, Zhou D (2019) lncRNA-D16366 Is a potential biomarker for diagnosis and prognosis of hepatocellular carcinoma. Med Sci Monit 25: 6581–6586.
- 12 Gu L, Liu J, Xu D, Lu Y (2019) Reciprocal feedback loop of the MALAT1-MicroRNA-194-YAP1 pathway regulates progression of acute pancreatitis. Med Sci Monit 25: 6894–6904.
- 13 Li J, Bu X, Chen X, Xiong P, Chen Z, et al. (2020) Predictive value of long non-coding RNA intersectin 1–2 for occurrence and in-hospital mortality of severe acute pancreatitis. J Clin Lab Anal 34: e23170.
- 14 Yu S, Li Y, Liao Z, Wang Z, Wang Z, et al. (2020) Plasma extracellular vesicle long RNA profiling identifies a diagnostic signature for the detection of pancreatic ductal adenocarcinoma. Gut 69: 540–550.
- 15 Liu H, Yu K, Ma P, Xiong L, Wang M, et al. (2019) Long noncoding RNA myocardial infarction-associated transcript regulated the pancreatic stellate cell activation to promote the fibrosis process of chronic pancreatitis. J Cell Biochem 120: 9547–9555.
- 16 Xu W, Zhou G, Wang H, Liu Y, Chen B, et al. (2020) Circulating lncRNA SNHG11 as a novel biomarker for early diagnosis and prognosis of colorectal cancer. Int J Cancer 146: 2901–2912.
- 17 Huang W, Huang F, Lei Z, Luo H (2020) LncRNA SNHG11 promotes proliferation, migration, apoptosis, and autophagy by regulating hsa-miR-184/AGO2 in HCC. Onco Targets Ther 13: 413–421.
- 18 Liu S, Yang N, Wang L, Wei B, Chen J, et al. (2020) lncRNA SNHG11 promotes lung cancer cell proliferation and migration via activation of Wnt/β-catenin signaling pathway. J Cell Physiol 235: 7541–7553.
- 19 Morikawa M, Derynck R, Miyazono K (2016) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol 8: a021873.
- 20 Sun L, Xiu M, Wang S, Brigstock DR, Li H, et al. (2018) Lipopolysaccharide enhances TGF-β1 signalling pathway and rat pancreatic fibrosis. J Cell Mol Med 22: 2346–2356.
- 21 Sanvito F, Nichols A, Herrera PL, Huarte J, Wohlwend A, et al. (1995) TGF-beta 1 overexpression in murine pancreas induces chronic pancreatitis and, together with TNF-alpha, triggers insulin-dependent diabetes. Biochem Biophys Res Commun 217: 1279–1286.
- 22 Ren YC, Zhao Q, He Y, Li B, Wu Z, et al. (2020) Legumain promotes fibrogenesis in chronic pancreatitis via activation of transforming growth factor β1. J Mol Med (Berl) 98: 863–874.
- 23 Novotny GE, Pau H (1984) Myofibroblast-like cells in human anterior capsular cataract. Virchows Arch A Pathol Anat Histopathol 404: 393–401.
- 24 Chen YT, Hsu H, Lin CC, Pan SY, Liu SY, et al. (2020) Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J Pathol 250: 55–66.
- 25 Zhang L, Zhang L, Huang Z, Xing R, Li X, et al. (2019) Increased HIF-1α in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev 2019: 6326517.
- 26 Liu Y, Feng W, Liu W, Kong X, Li L, et al. (2019) Circulating lncRNA ABHD11-AS1 serves as a biomarker for early pancreatic cancer diagnosis. J Cancer 10: 3746–3756.
- 27 Thomson DW, Dinger ME (2016) Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17: 272–283.
- 28 Zhang L, Wang L, Dong D, Wang Z, Ji W, et al. (2019) MiR-34b/c-5p and the neurokinin-1 receptor regulate breast cancer cell proliferation and apoptosis. Cell Prolif 52: e12527.
- 29 Cao Z, Zhang G, Xie C, Zhou Y (2019) MiR-34b regulates cervical cancer cell proliferation and apoptosis. Artif Cells Nanomed Biotechnol 47: 2042–2047.
- 30 Zhuang XF, Zhao LX, Guo SP, Wei S, Zhai JF, et al. (2019) miR-34b inhibits the migration/invasion and promotes apoptosis of non-small-cell lung cancer cells by YAF2. Eur Rev Med Pharmacol Sci 23: 2038–2046.
- 31 Liu C, Cheng H, Shi S, Cui X, Yang J, et al. (2013) MicroRNA-34b inhibits pancreatic cancer metastasis through repressing Smad3. Curr Mol Med 13: 467–478.
- 32 Guo X, Zhou Q, Su D, Luo Y, Fu Z, et al. (2020) Circular RNA circBFAR promotes the progression of pancreatic ductal adenocarcinoma via the miR-34b-5p/MET/Akt axis. Mol Cancer 19: 83.
- 33 Munding JB, Adai AT, Maghnouj A, Urbanik A, Zöllner H, et al. (2012) Global microRNA expression profiling of microdissected tissues identifies miR-135b as a novel biomarker for pancreatic ductal adenocarcinoma. Int J Cancer 131: E86–E95.
- 34 Chodorowska G, Głowacka A, Tomczyk M (2004) Leukemia inhibitory factor (LIF) and its biological activity. Ann Univ Mariae Curie Sklodowska Med 59: 189–193.
- 35 Shi Y, Gao W, Lytle NK, Huang P, Yuan X, et al. (2019) Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569: 131–135.
- 36 Hunter SA, McIntosh BJ, Shi Y, Sperberg RAP, Funatogawa C, et al. (2021) An engineered ligand trap inhibits leukemia inhibitory factor as pancreatic cancer treatment strategy. Commun Biol 4: 452.
- 37 Wrona E, Potemski P, Sclafani F, Borowiec M (2021) Leukemia inhibitory factor: a potential biomarker and therapeutic target in pancreatic cancer. Arch Immunol Ther Exp (Warsz) 69: 2.