ORIGINAL
Severe metabolic disorders coexisting with Werner syndrome: a case report
2021 Volume 68 Issue 3 Pages 261-267

Details
2021 Volume 68 Issue 3 Pages 261-267
Werner syndrome, also called adult progeria, is a heritable autosomal recessive human disorder characterized by the premature onset of numerous age-related diseases including juvenile cataracts, dyslipidemia, diabetes mellitus (DM), osteoporosis, atherosclerosis, and cancer. Werner syndrome is a segmental progeroid syndrome whose presentation resembles accelerated aging. The most common causes of death for WS patients are atherosclerosis and cancer. A 40-year-old female presented with short stature, bird-like facies, canities with alopecia, scleroderma-like skin changes, and non-healing foot ulcers. The patient reported a history of delayed puberty, abortion, hypertriglyceridemia, and juvenile cataracts. A clinical diagnosis of WS was made and subsequently confirmed. We discovered two WRN gene mutations in the patient, Variant 1 was the most common WRN mutation, nonsense mutation (c.1105C>T:p.R369Ter) in exon 9, which caused a premature termination codon (PTC) at position 369. Variant 2 was a frameshift mutation (c.1134delA:p.E379KfsTer5) in exon 9, which caused a PTC at position 383 and has no published reports describing. Patients with WS can show a wide variety of clinical and biological manifestations in endocrine-metabolic systems (DM, thyroid dysfunction, and hyperlipidemia). Doctors must be cognizant of early manifestations of WS and treatment options.
WERNER SYNDROME (WS) is a rare, autosomal recessive condition with multiple progeroid features caused by a mutation in the Werner gene (WRN) that encodes the protein WRN RecQ-like helicase [1, 2]. It was first described by Otto Werner in 1904 [3]. Classic WS is caused by WRN mutation and WS cases without WRN mutations is caused by LMNA mutation. WS patients suffer prematurely from a variety of age-related diseases in one or more of the body’s major systems (nervous, immune, connective tissue, and endocrine-metabolic systems), and die at an average age of 54 years [4]. In this article we report the results of genetic analysis in the diagnosis of a patient with WS.
A 40-year-old Chinese woman presented with a chief complaint of non-healing foot ulcers. After a thorough examination, the patient was diagnosed as having WS. The patient was healthy at birth and developed normally during childhood. There were sequentially-appearing clinical manifestations of WS in the patient: lack of a pubertal growth spurt (at age 14); canities with alopecia (at age 22); hoarseness (at age 25); skin atrophy (at age 28); cataracts (at age 33); and severe hypertriglyceridemia and DM (at age 34). She married at the age of 21 years and had 3 spontaneous abortions, each at 6 months of pregnancy. The patient had a normal menstrual history until the age of 36, afterwhich the menstrual cycle increased to 2 months, the menstrual period remained at 2–4 days, however the menstrual volume was reduced. The patient was being treated with Gansulin 30R® (66 U, 2x daily), but her fasting blood glucose (FPG) level and glycosylated hemoglobin (HbA1c) remained high (11.62 mmol/L and 8.7%, respectively) .
A clinical examination included blood pressure (130/70 mm Hg), heart rate (76 bpm), temperature (36.5°C), body weight (32 kg), height (147 cm), waist circumference (72 cm), body mass index (14.81 kg/m2), and assessment of physical features: bird-like facies; skin ulcers on the feet; abdominal obesity and thin limbs (Fig. 1A). The patient’s present father was not her biological father, her mother had lost contact with the patient’s biological father for more than 30 years. Clinical examinations of her mother, two half-brothers, nephews, and nieces revealed that all were in good health.
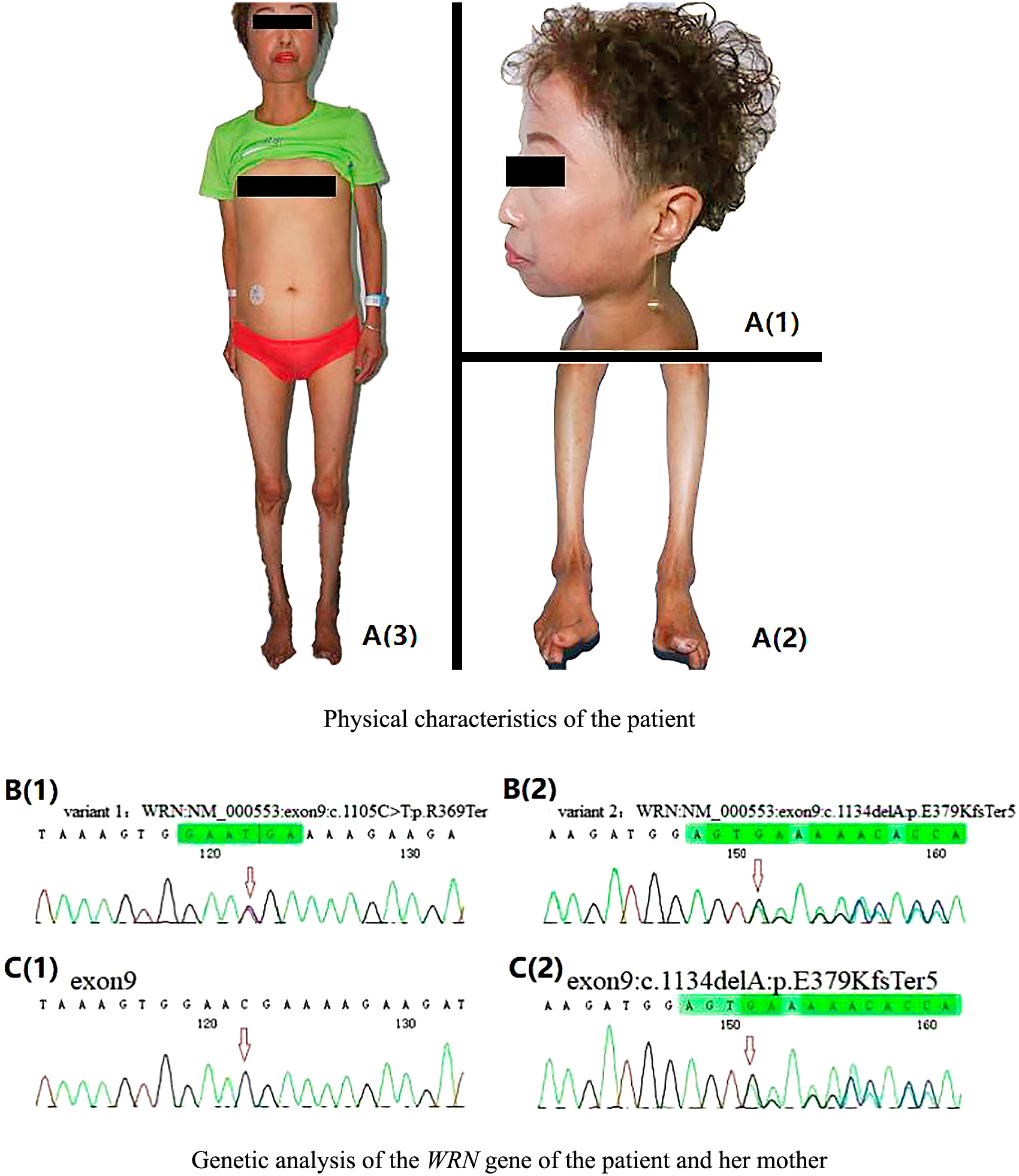
A: Physical characteristics of the patient. (A1) Bird-like facies. (A2) Skin ulcers on the feet. (A3) Abdominal obesity and thin limbs. B and C: Genetic analysis of the WRN gene of the patient and her mother. The mutated nucleotides are marked with an arrow. B. Patient DNA. B(1) Nonsense mutation c.1105 C>T in exon 9. B(2) Frame shift mutation c.1134delA in exon 9. C. Maternal DNA. C(1) No mutation in exon 9. C(2) Frame shift mutation c.1134delA in exon 9.
Laboratory tests revealed a decline in postprandial C-peptide levels and negative beta-cell autoantibodies (GADA, IAA, IA-2A, ICA, and ZnT8A), characteristic of Type 2 DM, poor glycemic control with concomitant severe hyperlipidemia, sub-clinical hypothyroidism with negative antithyroid peroxidase antibodies, and a slight decline in ovarian function. Visceral fat area measured by computed tomography, whole-body fat percentage and lean mass measured by dual-energy X-ray absorptiometry were 68 cm2, 27.9% and 21.92 kg. Routine laboratory including blood routine examination, routine urine test, liver function tests, renal function tests were all normal. Cardiovascular assessment including Electrocardiography (ECG) and echocardiography were normal. Ultrasound imaging revealed a fatty liver and hypoplastic uterus. The patient’s thyroid was of normal size with normal blood flow. Dual energy X-ray absorptiometry (DEXA) revealed osteopenia. Laboratory test data are presented in Table 1.
| Index | Results | Reference Range |
|---|---|---|
| Fasting plasma glucose (mmol/L) | 11.62 | 3.9–6.1 |
| Glycosylated hemoglobin (%) | 8.70 | 4.27–6.07 |
| Fasting serum C-peptide (ng/mL) | 1.77 | 0.5–3.8 |
| 1h postprandial serum C-peptide (ng/mL) | 1.98 | 5.38–6.67 |
| 2h postprandial serum C-peptide (ng/mL) | 1.69 | 4.10–5.56 |
| TG (mmol/L) | 24.56 | 0.56–1.71 |
| TC (mmol/L) | 6.28 | 2.90–5.17 |
| HDL-C (mmol/L) | 0.64 | 0.90–1.68 |
| LDL-C (mmol/L) | 1.16 | 2.70–3.40 |
| LH (mIU/mL) | 8.98 | 1.9–12.5 |
| FSH (mIU/mL) | 17.52 | 2.5–10.2 |
| PRL (ng/mL) | 43.35 | 2.8–29.2 |
| E2 (pg/mL) | 24.95 | 18.9–246.7 |
| PG (ng/mL) | 0.45 | 0.15–1.4 |
| T (ng/dL) | 8.76 | 9.01–47.94 |
| TT3 (nmol/L) | 1.92 | 0.92–2.79 |
| TT4 (nmol/L) | 91.7 | 58.1–140.6 |
| FT3 (pmol/L) | 4.34 | 3.5–6.5 |
| FT4 (pmol/L) | 10.27 | 11.5–22.7 |
| TSH (mU/L) | 7.445 | 0.55–4.78 |
| TPOAb (U/mL) | 35.1 | 0–60 |
| Tg (ng/mL) | 38.9 | 1.6–55 |
| TgAb (U/mL) | 28 | 0–60 |
TG, triglyceride; TC, total cholesterol; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein cholesterol; LH, luteinizing hormone; FSH, follicle-stimulating hormone; PRL, prolactin; E2, estradiol; PG, progesterone; T, testosterone; TSH, thyrotropin hormone; TPOAb, antithyroid peroxidase antibodies; Tg, thyroglobulin; TgAb, thyroglobulin antibody.
WS can be clinically diagnosed when the presence of multiple metabolic disorders and specific physical characteristics (as described above) are present based on the diagnostic criteria recommended by Takemoto et al. [5]. Genetic testing for the presence of a WRN mutation was pursued. Genetic analysis of the patient and her mother was performed with the approval of the Ethics Committee of The Second Hospital of Jilin University and the consent of the patient and her mother.
Whole genome sequencing identified two variants of the WRN gene in the patient’s genome (Fig. 1B). Variant 1 was a nonsense mutation c.1105C>T:p.R369Ter in exon 9, which caused the mutation of codon 369 from an arginine to a termination codon. Variant 1 has been recorded as a pathogenic mutation in the Human Gene Mutation Database (HGMD) and in the ClinVar database. Variant 2 was a frameshift mutation c.1134delA:p.E379KfsTer5 in exon 9, which caused the mutation of codon 379 from a glutamate to lysine and resulted in a termination codon at position 383. Genetic sequencing of the patient’s maternal genome revealed that her mother was a carrier of the exon 9: c.1134delA:p.E379KfsTer5 (Variant 2) mutation in a single allele (Fig. 1C).
Our patient was a non-obese female, without risk factors of metabolic diseases including type 2 DM and lipid disorders. The patient was noted to have specific physical characteristics (history of cataracts, hair changes, bird-like face, and low body mass index), which in addition to metabolic diseases, met diagnostic criteria for WS [5]. Genetic testing for WRN mutations confirmed our clinical diagnosis.
WS, an autosomal recessive premature ageing syndrome, is considered to be a model of natural human aging [1]. About 1,100 patients with WS have been reported between the first description by Otto Werner in 1904 [3] and 1994 [6]. People of Japanese descent account for 75% of all WS patients and 43% of WS patients are offspring of consanguineous marriages according to a Japanese nationwide epidemiological survey of WS conducted in 2009 [5, 7]. A study of the clinical features of 102 WS patients in Japan, revealed that the male-to-female ratio was 3:2 [8]. As long as the affected individual and his/her reproductive partner are non-consanguineous, offspring with WS is rare.
The clinical identification of WS as a genetic disorder led to the cloning of the responsible gene (WRN) in 1996 [9-11]. The protein defective in WS patients (WRN) is encoded by a member of the human RECQ gene family which contains both DNA exonuclease and helicase domains. WRN has been shown to participate in several DNA metabolic pathways, including DNA replication, recombination, repair, telomere maintenance, and transcription modulation [12]. Monnat RJ et al. have proposed a simple model for the pathogenesis of WS, in this model, lack of WRN protein causes genomic instability, accelerated telomere attrition and cell dysfunction and cell loss in many cell lineages with the accumulation of somatic mutations [13]. To date, a total of 83 different WRN mutations have been summarized, including eight previously unpublished mutations identified by the International Registry of Werner Syndrome (Seattle, WA) and the Japanese Werner Consortium (Chiba, Japan) [14]. WS patients primarily have the following WRN mutations; (a) nonsense mutations, which change amino acid codons to premature termination codons (PTCs), causing the premature termination of protein translation; (b) insertions and/or deletions, which lead to reading frameshift and premature termination of protein translation; (c) substitutions at splice junctions, which cause the skipping of exons and a subsequent frameshift; and (d) missense mutations, which cause amino acid change in the protein sequence [4]. The most frequent WS mutation is the Variant 1 nonsense mutation, c.1105C>T:p.R369Ter, in exon 9, which caused a PTC at position 369. This variant accounts for approximately 25% of all WS cases [4, 15]. Variant 2 was a frameshift mutation (c.1134delA:p.E379KfsTer5) in exon 9, which caused the mutation of codon 379 from a glutamate to a lysine and resulted in a PTC at position 383. There are no published reports describing Variant 2. We have not confirmed the presence of a Varient 2 protein but a possible mechanism that may eliminate its protein expression is nonsense-mediated decay (NMD) of mRNAs containing premature termination codons. NMD is a cellular surveillance system that prevents errors in gene expression. If generation of PTC mRNA is not eliminated through NMD, the abnormal proteins produced can be toxic to cells through dominant-negative or gain-of-function effects. There are three major ways that NMD can effect clinical outcomes: (1) changing the pattern of heredity, (2) causing different traits to be expressed from the same gene mutations, and (3) modifying the specific clinical phenotype [16].
WS patients usually develop normally in their childhood, but typically stop growing due to a lack of the pubertal growth spurt during the teen years. Most WS patients are reported to have short stature [4]. Median age of diagnosis ranges from late 30s to 40s [4, 5, 17]. The overall medical statistics (age, SD) for WS include: onset of growth delay (18.9, 7.7); canities (20.1, 10.4); scleroderma (26.4, 10.1); cataracts (31.2, 8.5); DM (31.5, 9.0); skin ulcers (34.7, 9.6); hypogonadism (35.6, 8.4); and osteoporosis (39.5, 7.55) [3]. Predictable subsequent events (age, SD) include: immune dysfunction (40.0, 10.5); atherosclerosis (40.6, 9.0); brain atrophy (40.7, 9.1); dementia (41.1, 7.7); malignancy (41.3, 9.2); and age at death (46.0, 11.6) [15]. We used “Werner syndrome” and “WRN mutation”, “Werner syndrome” and “LMNA mutation”, “Werner syndrome” or “progeria” to search in English database (Pubmed), and Chinese database (China National Knowledge Infrastructure (CNKI) up to 15th August 2020, and found 16 Chinese patients with WS reported [18-33]. Demographic (i.e., age, sex) and clinical characteristics of individual patients were summarized in Table 2.
| Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
Patient 5 |
Patient 6 |
Patient 7 |
Patient 8 |
Patient 9 |
Patient 10 |
Patient 11 |
Patient 12 |
Patient 13 |
Patient 14 |
Patient 15 |
Patient 16 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | F | F | M | F | F | M | F | M | M | F | M | F | M | M | M | M |
| Age at diagnosis | 46 | 26 | 45 | 18 | 43 | 31 | 30 | 45 | 39 | 18 | 41 | 19 | 26 | 37 | 31 | 38 |
| Age of initial symptoms | 24 | 18 | Early teens | 13 | Early teens | 13 | Early teens | Early teens | NM | 13 | 13 | 13 | 8 | NM | 14 | NM |
| Chief complaint | Foot ulcers | Alopecia | Foot ulcers | Dark skin | Alopecia Hyperglycemia | Foot ulcers | Loss of binocular vision | Atrioventricular block | Nerve deafness | Hyperglycemia | Arthralgia | Irregular menstruation, hyperglycemia | Loss of binocular vision | Foot ulcers | Foot ulcers | Foot ulcers |
| Initial presenting symptoms | Alopecia | Alopecia | Short stature | Dark skin | Short stature | Short stature | Short stature | NM | Sensorineural hearing loss | Short stature | Short stature | Short stature | Short stature | NM | Short stature | NM |
| Cataracts | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | No | No | Yes | No | Yes | Yes | Yes | No |
| DM | No | No | Yes | Yes | Yes | No | No | No | No | Yes | Yes | Yes | No | No | No | No |
| Hyperlipidemia | NM | No | NM | NM | Yes | NM | NM | NM | No | NM | NM | Yes | NM | NM | NM | NM |
| Osteoporosis | Yes | No | Yes | NM | Yes | NM | NM | NM | Yes | NM | Yes | NM | NM | NM | Yes | NM |
| Cancer | No | No | No | No | No | No | Yes | No | No | No | No | No | No | No | No | No |
| Arteriosclerosis | NM | No | Yes | NM | Yes | NM | NM | NM | No | NM | NM | NM | NM | Yes | NM | NM |
| Hypophrenia | No | Yes | No | No | No | No | Yes | Yes | No | No | No | No | NM | NM | NM | NM |
| Parental consanguinity | NM | No | Yes | No | NM | NM | NM | Yes | No | NM | Yes | NM | Yes | Yes | NM | Yes |
| Affected sibling | NM | No | Yes | No | NM | NM | NM | Yes | No | NM | NM | NM | NM | NM | NM | NM |
| Short stature | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | NM |
| Bird-like faces | NM | Yes | Yes | Yes | Yes | NM | Yes | NM | Yes | NM | Yes | Yes | Yes | Yes | NM | Yes |
| Hypogonadism | NM | Yes | NM | NM | Yes | Yes | NM | Yes | Yes | NM | Yes | Yes | NM | NM | Yes | NM |
| Senilism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | NM | Yes | Yes | Yes | Yes | Yes | NM |
| Voice change | NM | No | Yes | NM | NM | Yes | NM | Yes | Yes | NM | Yes | NM | NM | Yes | Yes | Yes |
| Gray hair or alopecia | Yes | Yes | Yes | Yes | Yes | NM | NM | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Scleroderma | Yes | Yes | Yes | Yes | Yes | Yes | Yes | NM | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Flat feet | NM | No | Yes | Yes | NM | NM | NM | NM | Yes | NM | NM | NM | NM | NM | NM | NM |
| Gene testing | No | No | No | No | No | No | No | No | 2967+237A>G 3309+26C>T c.2361G>T in Exon20 c.3237G>A in Exon27 |
L512R-(Leu512Ag) in the LMNA gene | c.2229_2230delAG | p.L140Q (c.419T>A) in the LMNA gene | c.3460_3461 insTTGTG) | (IVS28+2C) | 3250delG in exon 25 | (c.IVS28+2T>C) |
NM, not mentioned. Of the 16 patients listed, 8 were diagnosed as Werner syndrome by genetic testing, including 2 patients with atypical Werner syndrome (Patient 10 and Patient 12).
WS was accurately diagnosed in the patient through the collection of a detailed medical history, a careful physical examination, and obtaining extensive laboratory test results. The diagnosis of WS was based on the diagnostic criteria recommended by Takemoto et al. [5]. The treatment for the patient was comprehensive, including: lowering blood glucose and reducing insulin resistance (pioglitazone, 30 mg, once per day), lowering lipid levels (fenofibrate, 200 mg, once per day), antioxidant therapy (thioctic acid injection, 600 mg, once per day), neuroprotection (mecobalamin, 0.5 mg, 3 times per day), and proper care and dressing of foot ulcers and scleroderma.
Over the course of a 7-day treatment, FPG decreased to 8.0–9.0 mmol/L, the 2-hour postprandial glucose decreased to 6.0–11.0 mmol/L, the serum triglyceride level decreased from 24.56 to 19.85 mmol/L; the total serum cholesterol level decreased from 11.68 to 9.45 mmol/L, and the daily insulin requirement decreased from 66 to 38 U/day. Treatment after discharge included: Gansulin 30R® (38 U, 2x daily); pioglitazone (30 mg, once daily); fenofibrate (200 mg, once daily); and mecobalamin, (0.5 mg, 3x daily). The follow-up results after 6 months were: FPG (5.0–9.0 mmol/L); 2 hour postprandial glucose (7.0–13.0 mmol/L); serum triglyceride level (6.61 mmol/L); and total serum cholesterol level (4.37 mmol/L), The patient’s foot ulcers improved and the cooling sensation decreased.
When WS is diagnosed in patients, annual screening is important for early detection of tumors and cataracts. In addition, physicians should monitor any development of arteriosclerosis-related diseases. Treatments for the symptoms of WS include: surgery for refractory ulcers [34] and cataracts [31], anti-aging treatment such as vitamin C [35], or rapamycin [36], either with or without a farnesyltransferase inhibitor [37], and pluripotent stem cell therapy [38, 39]. With an increasing awareness of WS, the availability of genetic testing, and the variety of available investigative tools (genomics, epigenomics, transcriptomics, proteomics, and metabonomics), it is hoped that new approaches for the treatment of WS symptoms will also provide for the early recognition and treatment of age-related disorders [39].
We reported on a 40-year-old Chinese woman who presented with multiple metabolic disorders and progeria. She was clinically diagnosed with WS which was subsequently found to be caused by a WRN gene mutation in each allele. We discovered that the two WRN gene mutations included the Variant 1 nonsense mutation (c.1105C>T:p.R369Ter) in exon 9 (the most common WRN mutation) and a unique frameshift mutation (c.1134delA:p.E379KfsTer5) also in exon 9. Because there is currently no cure for WS, early diagnosis and treatment of WS will reduce, or eliminate, age-related disorders in WS patients. We hope that this case report will contribute to the clinical awareness and investigations of this rare disease.
The authors have nothing to disclose.
We thank the patient and appreciate the help of all family members who participated in this study.
We would also like to thank TopEdit (www.topeditsci.com) for English language editing of this manuscript.
This study involving Human Participants was approved by the ethical review committee of The Second Hospital of Jilin University (2020-014). All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
HL communicated with the patient and her family, acquired the clinical data, and drafted the initial manuscript. MY assisted with the writing and editing of the manuscript. HS assisted in evaluating the condition of the patient and performed literature searches to assist in the diagnosis of the disease. SW collected blood samples from the patient and her parents. HC designed and supervised the study and assisted in the revision and completion of the manuscript.