Abstract
HCP5 has been reported to be downregulated in ovarian granulosa cells (OGCs) and to facilitate cell proliferation. Human umbilical cord mesenchymal stem cell exosome (hucMSCs-exo) treatment can prevent OGCs apoptosis in vitro. However, the functional mechanism of HCP5 and hucMSCs-exo requires further exploration. Fluorescence-activated cell sorting (FACS) was performed to measure the expression of markers related to hucMSCs. The osteogenic and adipogenic potential of hucMSCs was measured by alkaline phosphatase (ALP) and Alizarin red and by oil red-O staining, respectively. Real-time quantitative polymerase chain reaction (RT–qPCR) and Western blotting were used to detect the mRNA and protein levels, respectively. Cell proliferation and apoptosis were measured by Cell Counting Kit-8 (CCK-8) assay, colony formation assay and flow cytometry. The interaction of HCP5/musashi RNA-binding protein 2 (MSI2) and oestrogen receptor alpha 1 (ESR1) mRNA was analysed using RNA pulldown and RIP assays. HucMSCs and exosomes were successfully isolated and identified. HucMSC-derived exosomes promoted the proliferation of OGCs and ESR1 expression and inhibited cell apoptosis. HCP5 overexpression in exosomes further enhanced these effects. MSI2 knockdown led to the opposite results. HCP5 targeted MSI2, and MSI2 knockdown reduced the decreases in HCP5 and ESR1 expression. Mechanistically, HCP5 in HucMSC-derived exosomes promoted ESR1 expression by binding to MSI2, which promoted the proliferation of OGCs.
THE INJURY to ovarian granulosa cells (OGCs) and subsequent apoptosis may be causes of premature ovarian failure (POF) induction. Therefore, it is urgent to explore the developmental mechanism of OGCs.
Human umbilical cord mesenchymal stem cells (hucMSCs) are ideal seed cells for tissue engineering due to their valuable potential for proliferation and differentiation, low immunogenicity, convenience of acquisition and safety [1]. Increasing numbers of studies have explored the application of hucMSCs in the treatment of different diseases. POF mice exhibited elevated oestradiol, decreased follicle-stimulating hormone, and significantly improved ovarian lesions after hucMSCs treatment [2]. Wang et al. demonstrated that hucMSCs transplantation reduced apoptosis of granulosa cells but promoted cell proliferation, which indicates that hucMSCs can enhance the release of cell growth factors and tissue regeneration [3]. Furthermore, another study reported that exosomes derived from hucMSCs (hucMSCs-exo) could integrate with injured OGCs, thereby inhibiting cell apoptosis in vitro [4]. The aim of this study was to explore how hucMSCs-exo affect the growth of OGCs.
Long noncoding RNAs (lncRNAs) and other contents are present in exosomes and can be transferred by exosomes to mediate communication between cells [5]. A previous study demonstrated that the lncRNA HCP5 is expressed at low levels in POF; furthermore, HCP5 is involved in the dysfunction of granulosa cells from patients with biochemical POF by regulating MSH5 transcription and DNA damage repair via targeting YB1 [6]. Moreover, HCP5 knockdown was reported to suppress the proliferation of KGN cells and to induce apoptosis [7]. Thus, the HCP5-related pathway has the potential to be a key factor to illustrate the growth of OGCs. Oestrogen receptor alpha 1 (ESR1) plays important roles in folliculogenesis, and ESR1 overexpression can stimulate the expression of oestrogen-related genes, regulate OGC proliferation and improve POF [8-10]. A previous study indicated that musashi RNA-binding protein 2 (MSI2), as an RNA-binding protein, regulates the stability of ESR1 mRNA and the protein expression of ESR1 in breast cancer [11]. Moreover, the bioinformatics prediction data suggested a binding site between HCP5 and MSI2. Although we reasonably suspect that the HCP5/ESR1/MSI2 pathway might affect OGCs growth, no relevant studies have been reported.
In this study, we investigated the interaction between MSI2 and HCP5 or ESR1 mRNA in OGCs for the first time and demonstrated that hucMSC-derived exosomes with HCP5 overexpression promoted the proliferation of OGCs but repressed apoptosis through the MSI2-ESR1 axis, suggesting that the investigation of the biological function of exosomal HCP5 is a promising topic.
Materials and Methods
Identification of hucMSCs and OGCs
HucMSCs, SVOG OGCs and KGN human ovarian granulose-like tumour cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM/F-12 (Gibco, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS; Gibco) and 100 U/mL penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C in a humidified incubator with 5% CO2. The morphology of hucMSCs was observed by microscopy.
Cells were washed in PBS containing 2% FBS, then were divided into several flow tubes and incubated with FITC-conjugated anti-human CD29, CD34, CD90, HLA-DR, HLA-ABC, PE-conjugated CD44, CD45, CD105, CD271, and APC-conjugated CD31, CD73, and CD133 (all purchased from Invitrogen) for 1 h at room temperature in the dark. The signals were analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA) using fluorescence-activated cell sorting (FACS) DIVA software (Version 6.1.2).
Alkaline phosphatase (ALP) and Alizarin red staining were used to determine osteogenic capability, while oil red O staining was performed to measure adipogenic potential. All osteogenic and adipogenic induction reagents were purchased from Gibco, and the experimental procedures were strictly performed in accordance with the manufacturer’s instructions.
Isolation and characterization of exosomes from hucMSCs
Exosomes derived from hucMSCs were isolated based on the protocol indicated on the exosome isolation kit (Invitrogen, USA). The culture medium of hucMSCs was centrifuged at 2,000 × g for 10 min at 4°C. Subsequently, the supernatant was centrifuged at 10,000 × g for 30 min at 4°C to filter debris, followed by centrifugation at 110,000 g for 70 min at 4°C to remove the supernatant. The precipitate was washed and resuspended in PBS. Purified exosomes were stored below –80°C for further experiments.
10 μL of exosomes was added to the carrier copper grid and then negatively stained 1 minute later in 30 μL phosphotungstic acid solution. After the copper grid was dried, the morphology of the exosomes was observed by transmission electron microscopy (TEM).
Exosomes (100 μL) were diluted in 1,000 μL of PBS and then added to a cuvette. After removing the air bubbles, the diameter of exosomes was recorded by dynamic light scattering (DLS).
The expression of the exosomal markers TFIIB, Lamin A/C, CD63, CD81, Hsp70, and TSG101 was measured by western blot.
Cell transfection
A specific plasmid was transfected into hucMSCs to obtain hucMSC-derived HCP5-overexpressing exosomes. KGN and SVOG cells were transfected with the HCP5, sh (short hairpin RNA) MSI2, shHCP5 and their corresponding negative control plasmids alone or in combination: hucMSCs-exo+HCP5, HCP5+shMSI2, shMSI2+hucMSCs-exo and the corresponding negative controls. All plasmids were synthesized by GenePharma (Shanghai, China). Cells were collected 48 h after transfection and then subjected to further experimentation.
RNA extraction and quantitative real-time PCR (qRT–PCR) assay
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen), and RNA quality was determined using a NanoDrop2000c (Thermo Scientific, Waltham, USA) according to the manufacturer’s instructions. Next, RNA was converted into cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo), and qRT–PCR was performed on an ABI 7900 system using SYBR Green Real-Time PCR master mix (Thermo). GAPDH served as an internal control. The sequences of all primers are shown as follows: forward,
5'-TATCCCTGTGAAGATGAACC-3' and reverse 5'-TGCCACCTCTAAATGTC CTA-3' for HCP5; forward, 5'- AGACGGACCAAAGCCACTTG-3' and reverse, 5' -CCCCGTGATGTAATACTTTTG-3' for ESR1; forward, 5'-GAAGGTCGGAGTCAACGGAT-3' and reverse, 5'-CCTGGAAGATGGTGATGGGAT-3' for GAPDH. Primers were synthesized by RiboBio (Guangzhou, China). The transcriptional level of targeted genes was analysed by the 2–ΔΔCt method.
Western blotting
Total protein was isolated using RIPA buffer (Beyotime Institute of Biotechnology, Haimen, China). After the protein concentration was quantified using a BCA Protein Assay Kit (Beyotime), the samples were separated by 10% SDS–PAGE, transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA) and blocked with 5% skim milk for 1 h at room temperature. Subsequently, the membranes were incubated overnight at 4°C with primary antibodies against TFIIB (1:5,000, ab109106, Abcam, Cambridge, UK), Lamin A/C (1:20,000, ab133256, Abcam), CD63 (1:1,000, ab217345, Abcam), CD81 (1:5,000, ab109201, Abcam), Hsp70 (1:1,000, ab181606, Abcam), TSG101 (1:2,000, ab83, Abcam), ESR1 (1:5,000, ab108398, Abcam), Cyclin D1 (1:20,000, ab134175, Abcam), CDK4 (1:5,000, ab108355, Abcam), Bcl-2 (1:1,000, ab59348, Abcam), Bax (1;5,000, ab32503, Abcam), MSI2 (1:2,000, ab76148, Abcam) and with their corresponding secondary antibodies for 1 h at 37°C. Finally, the protein bands were visualized by ECL reagent (Millipore); β-actin served as an internal control.
CCK-8 assay
Transfected KGN and SVOG cells were seeded into 96-well plates at a density of 5000 cells/well and cultured for 48 h. Next, 10 μL of CCK-8 solution was added to each well and the cells were incubated for 1 hour at 37°C. At 0, 12, 24, 48 and 72 h post-transfection, cell viability was detected at 490 nm by a spectrophotometer (BioRad, Hercules, CA, USA). The results represent the mean of three replicates under the same conditions.
Colony formation assay
Transfected cells (1 × 105) were seeded into 6-well plates and cultured in RPMI-1640 medium (Thermo) supplemented with 10% FBS and 100 U/mL penicillin (Thermo) at 37°C. The medium was changed every 2 days. After 14 days, the colonies were fixed in 4% paraformaldehyde and then stained with 0.1% crystal violet. A microscope was used to calculate the number of colonies.
Flow cytometry
According to the protocol of the Annexin V-FITC Apoptosis Detection Kit (Sigma–Aldrich; Merck KGaA, Darmstadt, Germany), the transfected cells were washed in binding buffer and then centrifuged at 500 × g for 5 min at room temperature. Next, the cell pellet was resuspended in binding buffer and successively incubated with 10 μL Annexin V-fluorescein isothiocyanate and 10 μL propidium iodide for 15 min in the dark at room temperature. A flow cytometer (BD Biosciences) was then used to analyse the results.
RNA pull-down
Biotin-labelled HCP5 and ESR1 mRNA or their antisense RNA was transcribed with Biotin RNA Labelling Mix and T7/SP6 RNA polymerase (Roche Diagnostics, Indianapolis, IN, USA) and then treated with RNase-free DNase I (Roche). After purification using an RNeasy Mini Kit (Roche), biotin-labelled RNAs were mixed with extracted KGN and SVOG cell nuclear proteins and then incubated with streptavidin agarose beads at room temperature for 1 h. The protein bands were visualized by silver staining followed by western blotting.
RNA immunoprecipitation (RIP) assay
The RIP assay was conducted using a Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore). KGN and SVOG cells were lysed in RIP lysis buffer, and then immunoglobulin G (anti-IgG) and argonaute 2 (anti-Ago2) antibodies were coated on the magnetic beads overnight. Then, the magnetic bead-bound complexes were immobilized with a magnet, and unbound materials were washed away. Part of the cell served as the negative control and was named Input. The coprecipitated RNA was extracted using TRIzolTM, and qRT–PCR was then used to analyse the purified RNA.
Statistical analysis
All experiments were performed with at least five biological replicates. All experimental data are expressed as the mean ± standard deviation (SD). Student’s t test was utilized to analyse the difference between two groups, and one-way analysis of variance with Tukey’s post-hoc test was performed to evaluate the differences among at least 3 groups using GraphPad (Ver. Prism 7, GraphPad Prism Software, La Jolla, CA, USA). A p value less than 0.05 was considered statistically significant.
Results
The collection and identification of hucMSCs and hucMSCs-exo
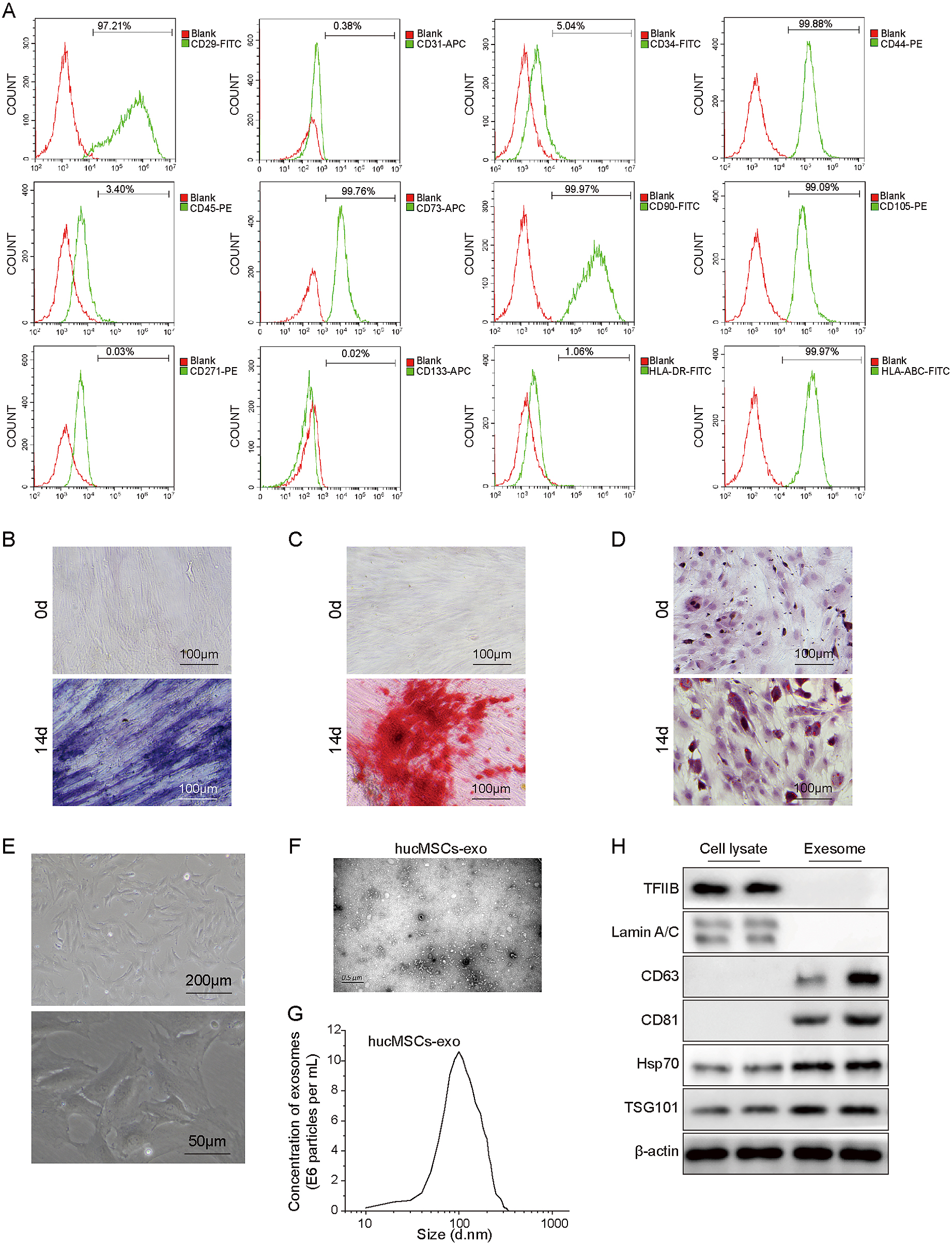
To identify isolated hucMSCs, we first measured the expression of positive biomarkers (CD29, CD44, CD73, CD90 and CD105), negative biomarkers (CD31, CD34, CD45, CD133 and CD271) and other surface markers (HLA-ABC and HLA-DR) of hucMSCs [12]. The results from fluorescence-activated cell sorting (FACS) suggested that positive markers and HLA-ABC were overexpressed in isolated hucMSCs, while negative markers and HLA-DR showed low expression (Fig. 1A). Next, ALP and Alizarin red staining were performed to determine the osteogenic potential of hucMSCs. We observed the staining images after osteogenic induction for 0 and 14 days, and the results indicated that ALP activity and Alizarin red were significantly higher at 14 days (Fig. 1B and 1C). After culturing the cells in adipogenic conditions for 14 days, we observed many lipid vacuoles by oil red O staining (Fig. 1D). Furthermore, as shown in Fig. 1E, the primary cultured hucMSCs were closely arranged and grew well and exhibited polygonal and star shapes. These findings suggested that we successfully isolated hucMSCs and that hucMSCs have osteogenic and adipogenic differentiation capabilities. Subsequently, the morphology of extracted hucMSCs-exo was visualized by transmission electron microscopy (TEM), and the exosome size was analysed by dynamic light scattering (DLS). The results demonstrated that the diameter of hucMSCs was approximately 100 nm (Fig. 1F and 1G). Western blotting analysis suggested that exosomes expressed the exosomal markers CD63 and CD81 but did not express TFIIB or Lamin A/C and demonstrated higher levels of Hsp70 and TSG101 than cell lysates (Fig. 1H). These data showed that hucMSCs-exo were successfully purified, and thus, they were used in subsequent experiments.
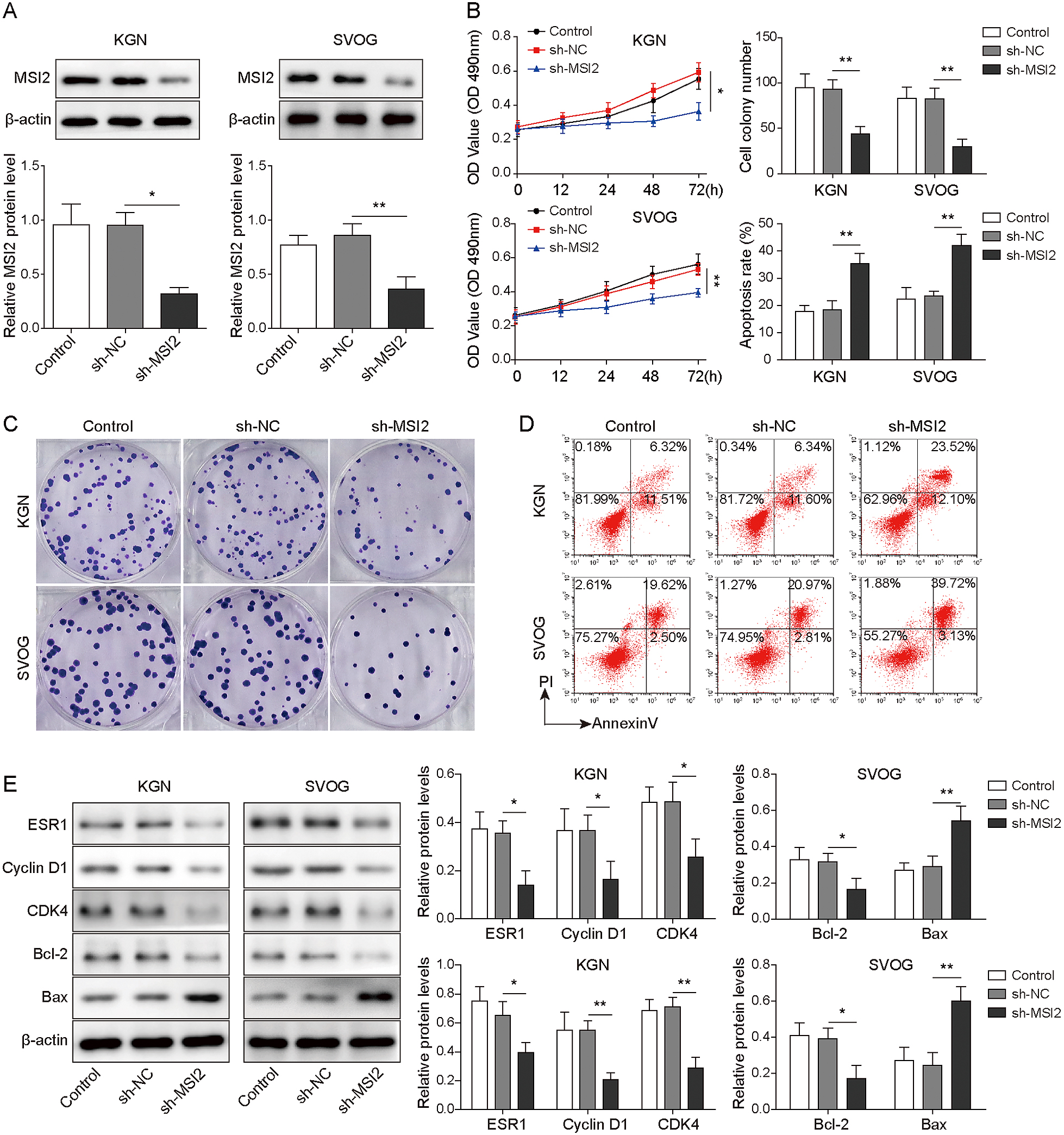
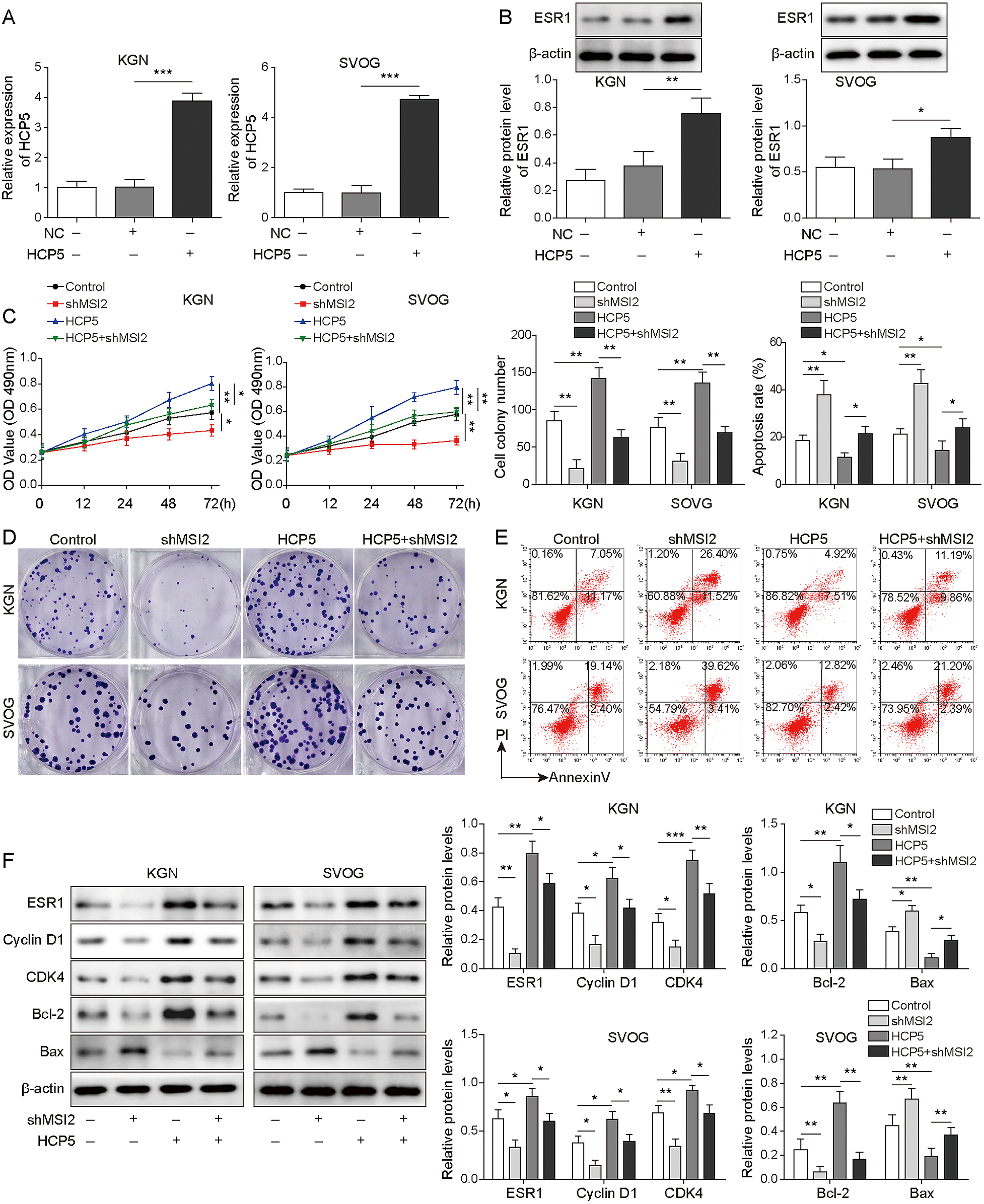
HCP5 has been reported to regulate the function of OGCs [13]. Here, the plasmid sh-HCP5 was successfully transfected into KGN and SVOG cells to knock down HCP5 expression (Fig. 2A). Next, cell viability, proliferation and apoptosis were analysed. The results indicated that HCP5 knockdown reduced the viability and the number of colony-forming cells but increased the apoptosis rate (Fig. 2B–D). Moreover, the expression of ESR1 and proliferation-related genes was measured. The expression of ESR1, Cyclin D1, CDK4 and Bcl-2 was decreased, while Bax expression was increased after silencing HCP5 (Fig. 2E). In conclusion, HCP5 contributes to the growth of KGN and SVOG cells.
MSI2 knockdown suppressed ESR1 expression and the proliferation of KGN and SVOG cells but promoted apoptosis
Considering that MSI2 regulates the mRNA stability and protein levels of ESR1, it is reasonable to suspect that MSI2 is involved in regulating OGCs growth through the HCP5/ESR1 pathway. First, we analysed the effect of MSI2 silencing on cell growth. After successfully culturing KGN and SVOG cells with low MSI2 expression (Fig. 3A), we determined the biological effects on the treated cells. We observed that cell viability and proliferation were reduced after MSI2 knockdown, while apoptosis was increased (Fig. 3B–3D). The results also indicated that the expression of ESR1, Cyclin D1, CDK4 and Bcl-2 was downregulated in KGN and SVOG cells transfected with shMSI2, while Bax levels were upregulated (Fig. 3E). Taken together, MSI2 impacts the proliferation and apoptosis of KGN and SVOG cells, which might be achieved through regulation of ESR1 levels. However, the specific mechanism remains to be further explored.
HCP5 increased the stability of ESR1 mRNA by combining with MSI2
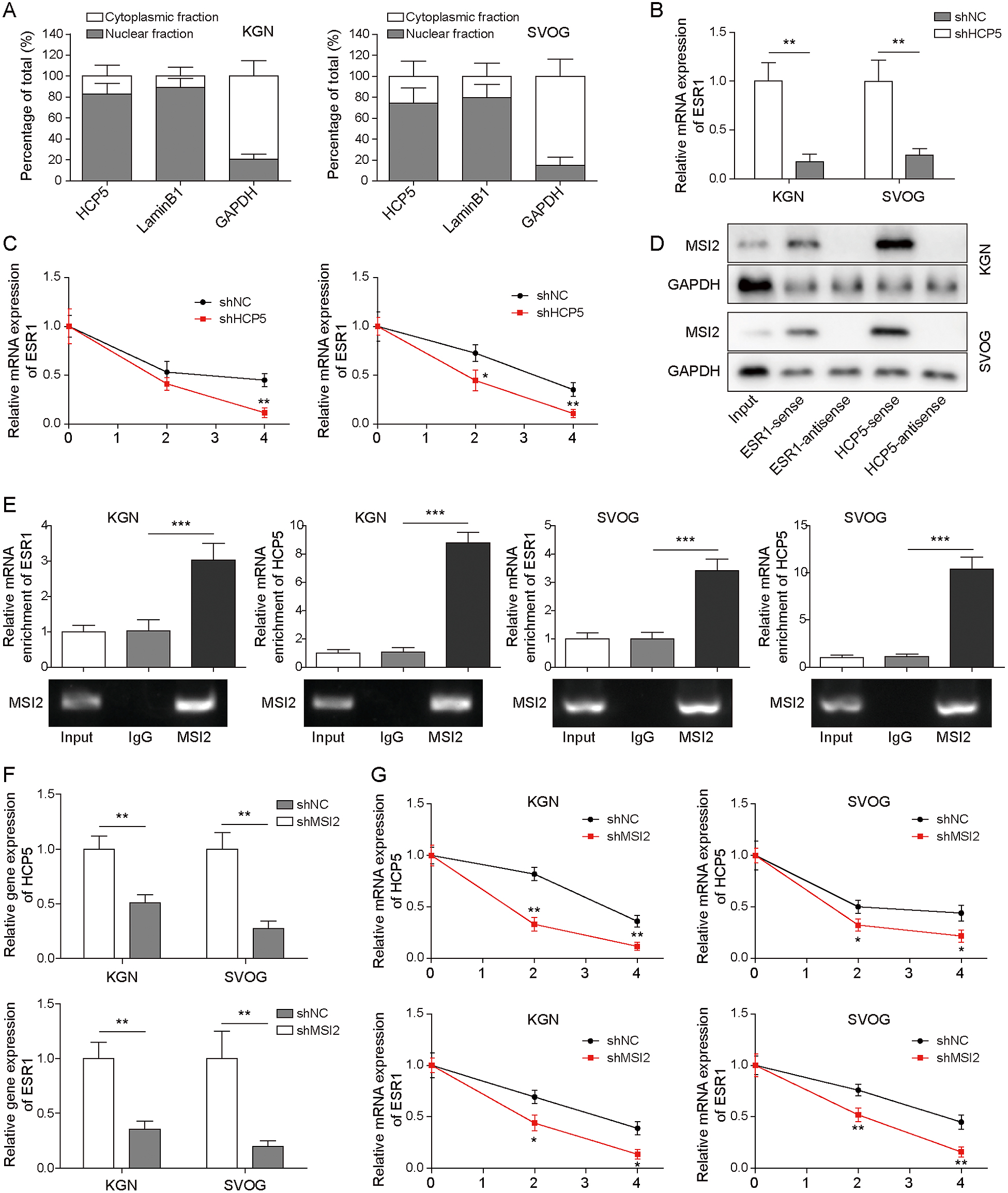
To further explore the mechanism by which HCP5 regulated ESR1 expression, we measured the distribution of HCP5 in the nucleus and cytoplasm. The results showed that HCP5 was mainly expressed in the cytoplasm, which indicated that HCP5 may upregulate ESR1 expression at the posttranscriptional level (Fig. 4A). Therefore, we then evaluated ESR1 mRNA expression and decay in KGN and SVOG cells after HCP5 knockdown. The results indicated that HCP5 silencing decreased ESR1 mRNA expression (Fig. 4B). Furthermore, after treatment with actinomycin D (ACD, 5 μg/mL), the half-life of ESR1 mRNA was shortened in cells transfected with shHCP5 (Fig. 4C). These data demonstrated that to some extent, HCP5 promotes ESR1 expression in OGCs by regulating the stability of ESR1 mRNA. Previous studies have shown that lncRNAs regulate downstream gene expression by targeting specific proteins [14], and as an RNA binding protein (RBP), MSI2 can interact with RNA molecules and affect RNA molecular stability and target transcription sequence translation. In addition, the bioinformatics prediction results indicated the presence of binding sites between HCP5/ESR1 mRNA and MSI2. Thus, RNA pull-down and RIP assays were performed to determine the existence of a regulatory relationship between them. As shown in Fig. 4D and 4E, MSI2 bound to sense HCP5 and ESR1 mRNA, and the abundance of MSI2 binding to HCP5 or ESR1 mRNA was much higher than that in the IgG group, which indicated that HCP5 directly binds to MSI2 or ESR1 mRNA in KGN and SVOG cells. Moreover, MSI2 knockdown resulted in low expression of HCP5 and ESR1 mRNA (Fig. 4F) and shortened the half-life of both mRNAs (Fig. 4G). In conclusion, these findings revealed a targeting relationship of HCP5/MSI2 and confirmed the enhancing effect of HCP5 on the interaction between ESR1 mRNA and MSI2.
After determining the interaction among HCP5/ESR1/MSI2 in vitro, we sought to verify whether this novel pathway could have a regulatory function in the mechanism of cell growth. As shown in Fig. 5A, KGN and SVOG cells transfected with HCP5 were successfully obtained. Then, we observed that HCP5 overexpression increased ESR1 levels (Fig. 5B). We further investigated whether HCP5 overexpression affected cell growth, and the results indicated that cell viability and proliferation were increased after HCP5 expression was enhanced, while apoptosis was inhibited. However, the effect of HCP5 overexpression on cell growth was reversed by cotransfection with shMSI2 (Fig. 5C–5E). Furthermore, HCP5 overexpression upregulated the expression of ESR1, Cyclin D1, CDK4 and Bcl-2 and downregulated Bax levels. However, these effects were reversed by MSI2 knockdown (Fig. 5F). Therefore, the HCP5/MSI2/ESR1 axis might be involved in the growth of KGN and SVOG cells.
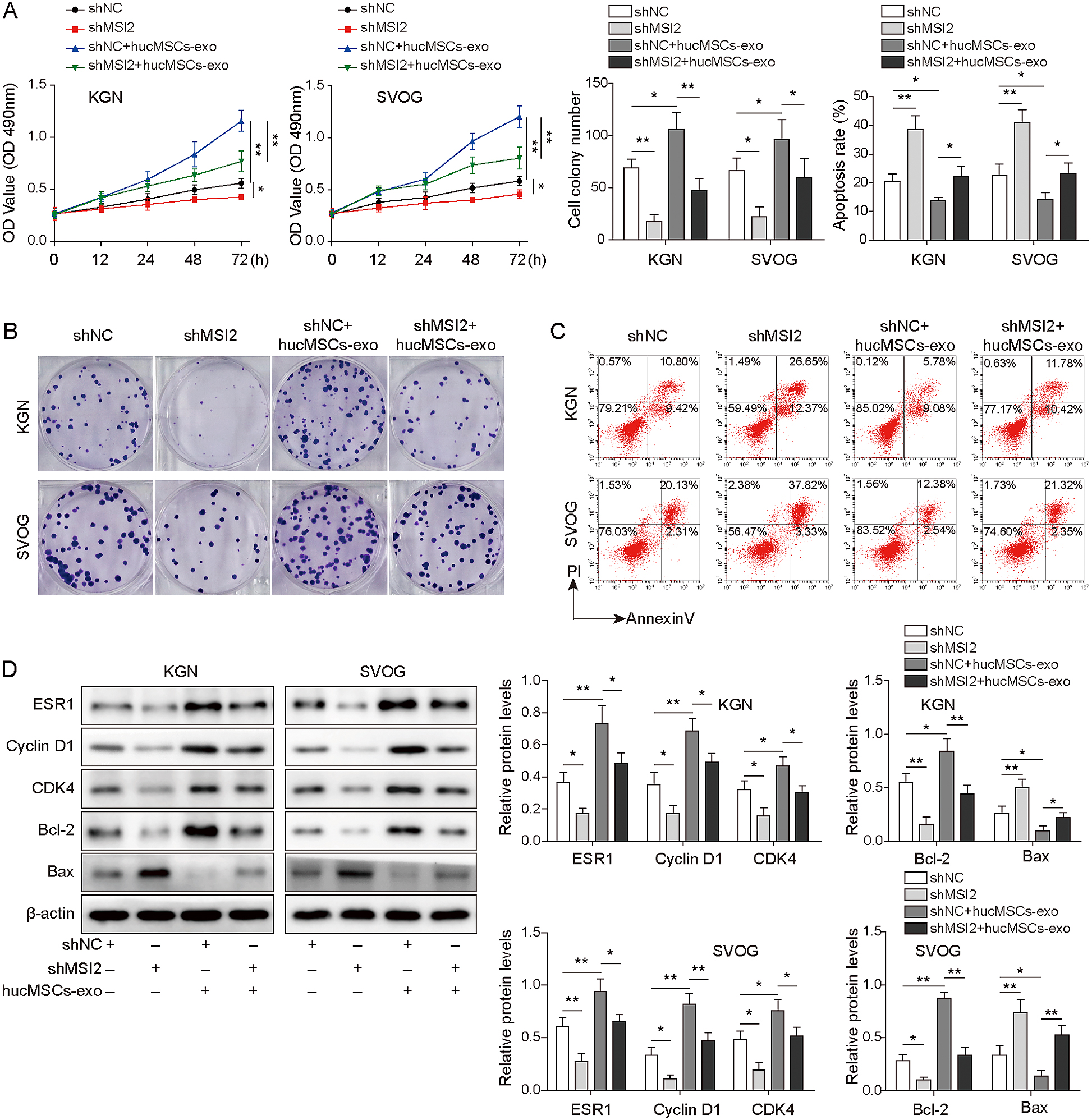
To explore the effect of hucMSCs-derived HCP5-overexpressing exosomes on KGN and SVOG cell growth, we first measured the efficiency of HCP5 overexpression. The results showed that HCP5 was successfully overexpressed (Fig. 6A). To determine whether KGN and SVOG cells could absorb exosomes, we labelled HCP5 with Cy3 and analysed the cells using laser scanning confocal microscopy. We observed that the labelled HCP5 in hucMSCs-exo was taken up by KGN and SVOG cells (Fig. 6B). As shown in Fig. 6C–6E, cell viability and proliferation was increased gradually with the addition of exosomes, while apoptosis was decreased. Interestingly, cell viability and proliferation were further enhanced, while apoptosis was further reduced in the hucMSCs-exo+HCP5 group. Subsequently, we found that the HCP5 levels of the hucMSCs-exo+NC and hucMSCs-exo+HCP5 groups increased successively in KGN and SVOG cells compared with those in the control group (Fig. 6F). Moreover, the expression of ESR1, Cyclin D1, CDK4 and Bcl-2 was increased and Bax expression was decreased after treatment with exosomes. As expected, co-transfection of KGN and SVOG cells with exosomes and HCP5 further enhanced these effects (Fig. 6G). In conclusion, hucMSCs-derived HCP5-overexpressing exosomes contribute to the growth of KGN and SVOG cells.
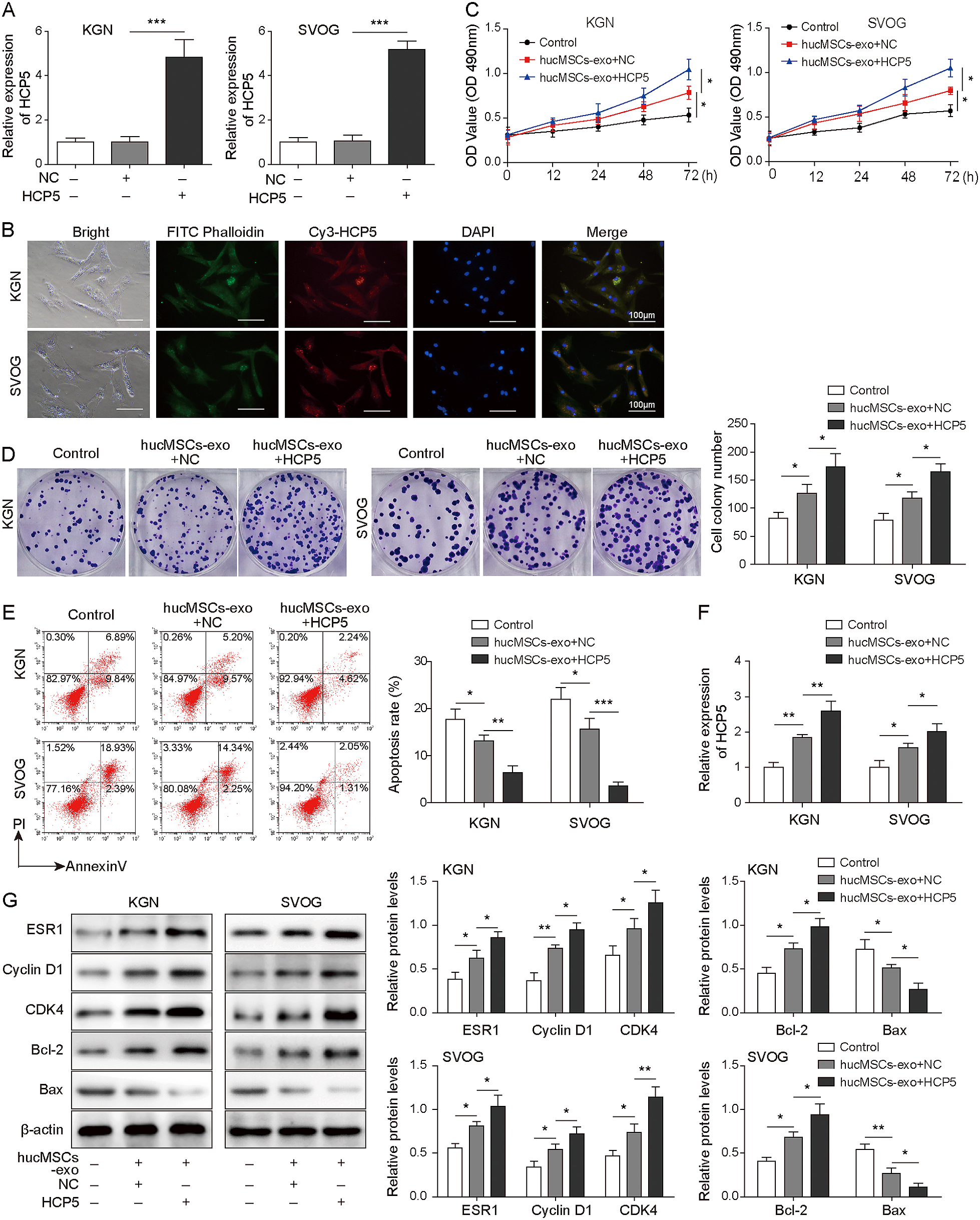
Next, KGN and SVOG cells transfected with shMSI2 were cultured with hucMSCs-exo and divided into the shNC, shMSI2, shNC+ hucMSCs-exo and shMSI2+ hucMSCs-exo groups. The results suggested that MSI2 knockdown reversed the promoting effect of hucMSCs-exo on cell proliferation and viability and the inhibitory effect on cell apoptosis (Fig. 7A–7C). Consistently, MSI2 silencing decreased the high expression of ESR1, Cyclin D1, CDK4 and Bcl-2 and increased the low expression of Bax, which was induced by transfection with hucMSCs-exo (Fig. 7D). These findings indicated that hucMSCs-exo-derived HCP5 affected cell growth by targeting MSI2, which upregulated ESR1.
Discussion
POF is a disastrous disease for women of childbearing age because it leads to complications, such as osteoporosis, and more seriously, infertility [15, 16]. The study of the growth mechanism of OGCs will provide a theoretical basis for the development of more effective treatment for POF. A previous study showed that hucMSCs-exo integrated with damaged OGCs and inhibited their apoptosis[4]. HucMSCs-exo can be absorbed by target cells and improve tissue damage by the horizontal transfer of lncRNAs, microRNAs and proteins [17]. In the present study, we found that hucMSCs-derived HCP5-overexpressing exosomes contributed to the growth of KGN and SVOG cells through the ESR1/MSI2 axis. We first isolated exosomes from hucMSCs, measured the diameter of exosomes using TEM and DLS, and analysed the expression of exosome markers to confirm that the extracted hucMSCs-exo could be used for subsequent experiments.
Laser scanning confocal microscopy was used to determine the effective absorption of hucMSCs-exo by OGCs, and we observed that Cy3-labelled HCP5 in hucMSCs-exo was present in OGCs, which was the basis of subsequent biological effects. HCP5 was demonstrated to be downregulated in POF and involved in the POF process by regulating dysfunctional granulosa cells [13]. Here, our findings not only indicated that transfected hucMSCs-exo played a promoting role in the growth of OGCs and ESR1 expression, but also, more importantly, that hucMSCs-exo-derived HCP5 overexpression further enhanced the effect. The loss of granulosa cells could lead to follicle atresia, which results in ovarian insufficiency and ultimately causes POF in vitro [18-20]. Therefore, the positive effect of HCP5 on cell growth shows possible therapeutic potential. Furthermore, we observed that MSI2 knockdown could inhibit the proliferation of OGCs and the expression of ESR1. MSI2 is an RNA binding protein that can coordinate the life cycle of mRNA and determine stability and translation efficiency [21]. MSI2 plays a key role in the functioning of mammalian stem cells and has been shown to be involved in the pathogenesis of different cancers [22], such as prostate and liver cancers [23, 24]. However, the function of MSI2 in OGCs growth has not been reported. Our data confirmed for the first time that MSI2 regulated the growth of OGCs. Combined with the effect of HCP5 on OGCs, we speculated that HCP5 might regulate the proliferation and apoptosis of KGN and SVOG cells by interacting with MSI2.
MSI2 has been demonstrated to be a regulator of mRNA processing and translation, as it regulates RNA splicing, polyadenylation, stability and degradation [25]. For example, MSI2 overexpression in breast cancer increased the stability of the ESR1 protein by binding to ESR1 mRNA [11]. Consistently, we found that MSI2 interacted with ESR1 mRNA in OGCs. In addition, we observed a binding site between HCP5 and MSI2, which could form a complex. MSI2 knockdown significantly reduced the binding of HCP5 and ESR1 mRNA. Several previous studies have also reported that lncRNAs affect the development of multiple diseases by regulating MSI2 expression [26]. For example, the lncRNA DANCR upregulates MSI2 levels by sponging miR-149 and promoting malignant phenotypes of bladder cancer [27]. Here, the results of functional experiments indicated that MSI2 knockdown could reverse the promoting effect of hucMSCs-exo-derived HCP5 overexpression on the growth of OGCs, indicating that hucMSCs-exo-derived HCP5 affected OGCs growth by upregulating ESR1 via targeting MSI2 in vitro.
In conclusion, our findings demonstrated for the first time that HCP5 increased the stability of ESR1 mRNA and ESR1 levels by binding to MSI2, thus promoting the proliferation of OGCs while inhibiting apoptosis. These observations provide a feasible theoretical basis for the development of new therapeutic approaches, which warrant further investigation.
Acknowledgments
This work was supported by the Hainan Province Science and Technology Special Fund (ZDYF2020131) and Hainan Province Clinical Medical Center.
Disclosure
The authors have no commercial or other associations that might pose a conflict of interest.
Authorship
Yu-Ting Suni: experimental studies, data acquisition, writing- original draft preparation. Jun-Hong Cai: data analysis, writing- reviewing and editing. Shan Bao: concepts, design, funding acquisition, supervision. All the authors approved for the final version.
References
- 1 Li T, Xia M, Gao Y, Chen Y, Xu Y (2015) Human umbilical cord mesenchymal stem cells: an overview of their potential in cell-based therapy. Expert Opin Biol Ther 15: 1293–1306.
- 2 Shen J, Cao D, Sun JL (2020) Ability of human umbilical cord mesenchymal stem cells to repair chemotherapy-induced premature ovarian failure. World J Stem Cells 12: 277–287.
- 3 Wang Z, Wei Q, Wang H, Han L, Dai H, et al. (2020) Mesenchymal stem cell therapy using human umbilical cord in a rat model of autoimmune-induced premature ovarian failure. Stem Cells Int 2020: 3249495.
- 4 Sun L, Li D, Song K, Wei J, Yao S, et al. (2017) Exosomes derived from human umbilical cord mesenchymal stem cells protect against cisplatin-induced ovarian granulosa cell stress and apoptosis in vitro. Sci Rep 7: 2552.
- 5 Wahlgren J, Statello L, Skogberg G, Telemo E, Valadi H (2016) Delivery of small interfering RNAs to cells via Exosomes. Methods Mol Biol 1364: 105–125.
- 6 Zhai X, Wu Y, Wang Z, Zhao D, Li H, et al. (2020) Long noncoding RNA LINC01133 promotes the malignant behaviors of renal cell carcinoma by regulating the miR-30b-5p/Rab3D axis. Cell Transplant 29: 963689720964413.
- 7 Chen Y, Zhang X, An Y, Liu B, Lu M (2020) LncRNA HCP5 promotes cell proliferation and inhibits apoptosis via miR-27a-3p/IGF-1 axis in human granulosa-like tumor cell line KGN. Mol Cell Endocrinol 503: 110697.
- 8 Kim S, Pyun JA, Cha DH, Ko JJ, Kwack K (2011) Epistasis between FSHR and CYP19A1 polymorphisms is associated with premature ovarian failure. Fertil Steril 95: 2585–2588.
- 9 Yoon SH, Choi YM, Hong MA, Lee GH, Kim JJ, et al. (2010) Estrogen receptor α gene polymorphisms in patients with idiopathic premature ovarian failure. Hum Reprod 25: 283–287.
- 10 Bretherick KL, Hanna CW, Currie LM, Fluker MR, Hammond GL, et al. (2008) Estrogen receptor alpha gene polymorphisms are associated with idiopathic premature ovarian failure. Fertil Steril 89: 318–324.
- 11 Kang MH, Jeong KJ, Kim WY, Lee HJ, Gong G, et al. (2017) Musashi RNA-binding protein 2 regulates estrogen receptor 1 function in breast cancer. Oncogene 36: 1745–1752.
- 12 Mafi P, Hindocha S, Mafi R, Griffin M, Khan WS (2011) Adult mesenchymal stem cells and cell surface characterization—a systematic review of the literature. Open Orthop J 5: 253–260.
- 13 Wang X, Zhang X, Dang Y, Li D, Lu G, et al. (2020) Long noncoding RNA HCP5 participates in premature ovarian insufficiency by transcriptionally regulating MSH5 and DNA damage repair via YB1. Nucleic Acids Res 48: 4480–4491.
- 14 Zhu J, Fu H, Wu Y, Zheng X (2013) Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci 56: 876–885.
- 15 Beck-Peccoz P, Persani L (2006) Premature ovarian failure. Orphanet J Rare Dis 1: 9.
- 16 Vujovic S, Ivovic M, Tancic-Gajic M, Marina L, Barac M, et al. (2012) Premature ovarian failure. Srp Arh Celok Lek 140: 806–811.
- 17 Herrera MB, Fonsato V, Gatti S, Deregibus MC, Sordi A, et al. (2010) Human liver stem cell-derived microvesicles accelerate hepatic regeneration in hepatectomized rats. J Cell Mol Med 14: 1605–1618.
- 18 Gao F, Zhang J, Wang X, Yang J, Chen D, et al. (2014) Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum Mol Genet 23: 333–341.
- 19 Sun Z, Zhang H, Wang X, Wang QC, Zhang C, et al. (2018) TMCO1 is essential for ovarian follicle development by regulating ER Ca 2+ store of granulosa cells. Cell Death Differ 25: 1686–1701.
- 20 Yeung CK, Wang G, Yao Y, Liang J, Tenny Chung CY, et al. (2017) BRE modulates granulosa cell death to affect ovarian follicle development and atresia in the mouse. Cell Death Dis 8: e2697.
- 21 Anji A, Kumari M (2016) Guardian of genetic messenger-RNA-binding proteins. Biomolecules 6: 4.
- 22 Okano H, Imai T, Okabe M (2002) Musashi: a translational regulator of cell fate. J Cell Sci 115: 1355–1359.
- 23 Zhao J, Zhang Y, Liu XS, Zhu FM, Xie F, et al. (2020) RNA-binding protein Musashi2 stabilizing androgen receptor drives prostate cancer progression. Cancer Sci 111: 369–382.
- 24 Wang X, Wang R, Bai S, Xiong S, Li Y, et al. (2019) Musashi2 contributes to the maintenance of CD44v6+ liver cancer stem cells via notch1 signaling pathway. J Exp Clin Cancer Res 38: 505.
- 25 Pereira B, Billaud M, Almeida R (2017) RNA-binding proteins in cancer: old players and new actors. Trends Cancer 3: 506–528.
- 26 Wang ZL, Wang C, Liu W, Ai ZL (2019) Emerging roles of the long non-coding RNA 01296/microRNA-143-3p/MSI2 axis in development of thyroid cancer. Biosci Rep 39: BSR20182376.
- 27 Zhan Y, Chen Z, Li Y, He A, He S, et al. (2018) Long non-coding RNA DANCR promotes malignant phenotypes of bladder cancer cells by modulating the miR-149/MSI2 axis as a ceRNA. J Exp Clin Cancer Res 37: 273.