Abstract
Hypertriglyceridemia is caused not only by environmental factors but also by genetic factors. Severe hypertriglyceridemia is prone to complications of acute pancreatitis. Here, we report a whole-exome sequencing (WES) analysis for a young hypertriglyceridemic patient with recurrent acute pancreatitis and the patient’s mother. A 28-year-old hypertriglyceridemic female was admitted to our hospital. At 23 years old, a health checkup clarified her hypertriglyceridemia. At the age of 26 and 27, she had repeated acute pancreatitis with severe hypertriglyceridemia (serum triglyceride level were 3,888 mg/dL and 12,080 mg/dL, respectively). The patient’s BMI was 29.0 kg/m2, and blood samples under fibrate medication showed triglyceride 451 mg/dL and HbA1c 7.2%. Type V dyslipidemia became more apparent at postprandial state. The WES analysis showed that the patients had two heterozygous variants in Apolipoprotein A5 (APOA5) gene (p.G185C and p.V153M), a heterozygous variant in Apolipoprotein E (APOE) gene (p.R176C), three heterozygous variants in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene (p.T1220I, p.R1453W and p.V470M). On the other hand, her mother, who had moderate hypertriglyceridemia without acute pancreatitis, had a heterozygous variant in APOA5 gene (p.G185C) and two heterozygous variants in CFTR gene (p.T1220I and p.V470M). These results suggest that the more severe pathology of the patient than her mother might be due to the possible compound heterozygous APOA5 variants, the heterozygous APOE variant, and the possible compound heterozygous CFTR variants. In this case, WES analyses were useful to evaluate not only the causative genes of hypertriglyceridemia (APOA5 and APOE) but also the genes involved in the development of acute pancreatitis (CFTR) simultaneously.
HYPERTRIGLYCERIDEMIA is a risk factor for atherosclerosis, and severe hypertriglyceridemia, which is characterized by serum triglyceride (TG) levels ≥1,000 mg/dL, is prone to complications of acute pancreatitis [1-4]. However, it was reported that patients who suffered from acute pancreatitis are only about 20% in patients with serum TG of 1,000 mg/dL or more [5]. What kind of patients, who are at high risk for acute pancreatitis, has not been clear.
Hypertriglyceridemia is caused not only by diabetes mellitus, obesity, and environmental factors (excessive alcohol consumption, excessive carbohydrate, and fat intake, etc.) but also by genetic factors. Recently, the development of next-generation sequencing technology has enabled extensive genome analysis, which had revealed the association of monogenic hypertriglyceridemia with genes such as lipoprotein lipase (LPL), apolipoprotein A5 (APOA5), apolipoprotein C-II (APOC2), lipase maturation factor 1 (LMF1), glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1 (GPIHBP1), and apolipoprotein E (APOE) [6, 7]. However, as far as we have searched, there is no report that, using a comprehensive genetic approach, investigated causative genes of severe hypertriglyceridemia and its complication such as acute pancreatitis.
In this case report, whole-exome sequencing (WES) analysis was performed for a young severe hypertriglyceridemic patient with repeated acute pancreatitis and her mother with moderate hypertriglyceridemia and no history of acute pancreatitis, to investigate the genetic predisposition to hypertriglyceridemia and acute pancreatitis.
Case Presentation
The patient is a 28-year-old Japanese female who was referred to our hospital for a thorough examination of hypertriglyceridemia. From 20 to 23 years old, she was treated daily with oral steroids for refractory urticaria. During that period, her weight has increased from 58 kg to 79 kg (at 23 years old). At the age of 23, a health checkup clarified high TG levels (300–400 mg/dL), but she did not visit hospital. At the age of 26, she suffered from severe acute pancreatitis, and a blood test revealed severe high TG (3,888 mg/dL). At that time, her HbA1c was as high as 8.3%, and she was first diagnosed with diabetes mellitus. She thus started to receive medication with bezafibrate, nicotinic acid, SGLT2 inhibitor and injections of long-acting insulin, until her admission. After that, her TG levels were not well controlled, for example, 784 mg/dL and 1,424 mg/dL, at which time her HbA1c was 7.8% and 7.9%. At 27 years of age, she suffered from acute pancreatitis again without any apparent triggers, such as heavy drinking or overeating. At that time, her blood samples showed a high TG level of 12,080 mg/dL. When she was 28 years old, she was referred to our hospital for a thorough examination and was admitted. On the day of hospitalization, long-acting insulin (8 unit/day) was stopped. She had no drinking or smoking habits and had not been taking steroidal medication for at least a year prior to her hospitalization. Family history showed that her mother had hypertriglyceridemia (Supplemental Fig. 1). There were no consanguineous marriages or premature cardiovascular events in the family. Physical examination on admission was as follows: height 158.8 cm, weight 73.1 kg, BMI 29.0 kg/m2, waist circumference 93 cm, visceral fat area 61.7 cm2, pulse rate 103/min (regular rhythm), and blood pressure 115/63 mmHg. She had no eruptive xanthoma. Table 1 shows the patient’s blood sampling findings on admission: HbA1c 7.2%, fasting blood glucose 116 mg/dL, T-chol 158 mg/dL, TG 451 mg/dL, HDL-chol 39 mg/dL and LDL-chol 54 mg/dL. Secondary cause of hypertriglyceridemia such as Cushing syndrome was ruled out. The carotid artery’s doppler ultrasound showed no atherosclerotic changes and plaques (max IMT: rt 0.59 mm/lt 0.53 mm). Abdominal CT showed fatty liver and gallstones.
Table 1
Results of Laboratory Tests on Admission
| 【Hematological Data】 |
|
【Urinalysis】 |
|
| WBC |
4,250/μL |
Specifity gravity |
1.043 |
| RBC |
476 × 104/μL |
pH |
5.5 |
| Hb |
13.6 g/dL |
Protein |
(–) |
| Plt |
22.7 × 104/μL |
Glucose |
(4+) |
|
|
Occult blood |
(–) |
| 【Biochemical Data】 |
|
Cortisol |
39 μg/day |
| Na |
139 mEq/L |
|
|
| K |
3.7 mEq/L |
【Endocrine Data】 |
|
| Cr |
0.48 mg/dL |
TSH |
2.17 μU/mL |
| AST |
11 U/L |
FT4 |
1.1 ng/dL |
| ALT |
8 U/L |
FT3 |
2.6 pg/mL |
| Alb |
4.0 g/dL |
ACTH |
25 pg/mL |
| T-chol |
158 mg/dL |
Cortisol |
25.4 mg/dL |
| TG |
451 mg/dL |
|
|
| HDL-chol |
39 mg/dL |
【Serological Data】 |
|
| LDL-chol |
54 mg/dL |
IgG |
941 mg/dL |
| FPG |
116 mg/dL |
IgA |
258 mg/dL |
| HbA1c |
7.2% |
IgM |
142 mg/dL |
| F-IRI |
12.9 μU/mL |
ANA |
(–) |
WBC: white blood cells, RBC: red blood cells, Hb: hemoglobin, Plt: platelets, Na: sodium, K: potassium, Cr: creatinine, AST: aspartate aminotransferase, ALT: alanine aminotransferase, Alb: albumin, T-chol: total cholesterol, TG; triglyceride, HDL-chol: high-density lipoprotein cholesterol, LDL-chol: low-density lipoprotein cholesterol, FPG:fasting plasma glucose, HbA1c: Hemoglobin A1c, F-IRI: fasting immuno-reactive insulin, TSH: thyroid stimulating hormone, FT4: free thyroxine, FT3: free triiodothyronine, ACTH: adrenocorticotropic hormone, ANA: anti-nuclear antibody
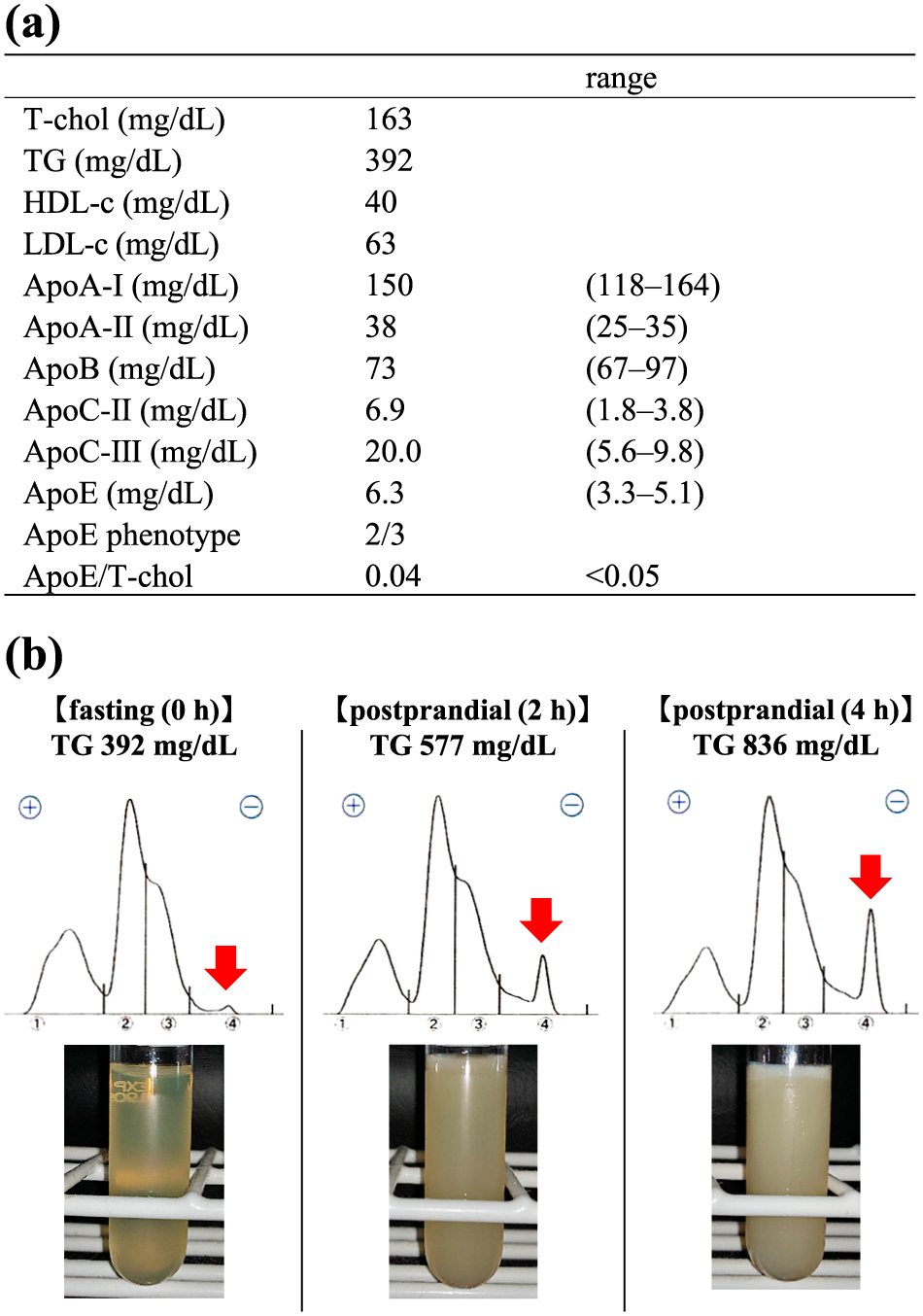
On day 15 after admission, the lipid profile was re-evaluated after glycemic control with a lipid-restricted diet (total 1,400 kcal, 35 g fat, 60 g protein per day) under fibrate (bezafibrate 400 mg/day) and SGLT2 inhibitor (empagliflozin 25 mg/day). The fasting serum TG level remained high at 392 mg/dL. There was no elevation of total cholesterol level (T-chol 163 mg/dL), ApoE/T-chol ratio was less than 0.05 (ApoE 6.3 mg/dL; range: 3.3–5.1), ApoE phenotype was 2/3. And so, familial type III hyperlipidemia was ruled out (Fig. 1a). Fasting lipoprotein electrophoresis (agarose gel) showed high preβ (suggesting very low density lipoprotein (VLDL) and only a small amount of origin residue (suggesting chylomicrons). After standing still of the serum at 4°C (the refrigerator test), the serum appearance was cloudy with only a small cream layer (Fig. 1b). These data suggested to be Type V dyslipidemia. After 2 hours and 4 hours postprandial, the origin residue and cream layer increased over time, indicating increasing chylomicrons (Fig. 1b). The amount of LPL protein before the heparin-loading test was 45 mg/dL (TG 526 mg/dL, serum free fatty acid 792 μEq/L (normal range; 140–850)). After heparin administration (30 units/kg), the LPL protein amount was 238 mg/dL, which seemed within the standard range (TG 354 mg/dL, serum free fatty acid 5,965 μEq/L) [8]. Furthermore, LPL and hepatic lipase (HL) activities were analyzed, and the saline method showed LPL activity of 9.6 μmol/L/h (standard range; 6.76 ± 2.56) and HL activity of 6.0 μmol/L/h (standard range: 6.06 ± 2.56), both within the standard range [9]. On day 23 after admission, the patient was discharged with TG 406 mg/dL under bezafibrate (400 mg/day) and body weight of 71.2 kg. After that, her TG was shown 114 mg/dL and 330 mg/dL in the outpatient department.
The patient had diabetes mellitus and obesity, which are risk factors for elevated triglycerides, but she does not have a heavy drinking habit. Even if considering her mild obesity (BMI 29.0 kg/m2) and diabetes mellitus (HbA1c 7.2%) for severe hypertriglyceridemia, her recurrent episodes of acute pancreatitis suggested the possibility of genetic factors being involved in her hypertriglyceridemia and pancreatitis. Therefore, we performed a WES analysis for the patient. In addition, to evaluate more the significance of detected gene variants in the patients, we performed WES analysis for her mother, having moderate hypertriglyceridemia with fasting TG around 500 mg/dL. The mother had no diabetes, no history of pancreatitis, but heavy alcohol drinking. We performed human genome sequence analyses after obtaining written and signed informed consent from the patient and her mother. Osaka University Research Ethics Committee approved this study procedure on 1 June 2016 (approval number: 702). The patient and her mother agreed to participate in the study and provided verbal informed consent to publish this report. Her father was divorced from her mother and was not available for genetic analysis at the time of the study.
Blood samples were collected from the patient and her mother. Genomic DNA was extracted from the whole venous blood using QIAamp DNA Blood Minikit (Qiagen, Hilden, Germany). WES was performed using the Agilent SureSelect Human All Exon V6 (Agilent Technologies, Santa Clara, CA) and paired-end 100 bp reads using the Illumina HiSeq 3000 platform (Illumina, Inc. San Diego, CA). Image analysis, base calling, and demultiplexing were performed using the Illumina bcl2fastq2 conversion software v2.20. FASTQ files were quality checked using FASTQC, and low-quality reads were removed using trimmomatic-0.36. Read alignment was performed using standard parameterized BWA v0.7.17 for human genome assembly hg19 (GRCh37). SNVs or short In/dels were called according to GATK best practice (GATK4.0.3). Called variants are filtered using GATK Variant Filtration, and variants that meet conditions that “QD <2.0, FS >60.0, MQ <40.0, MQRankSum <–12.5, ReadPosRankSum <–8.0, SQR >4.0” are analyzed. Then, annotation information was added to the obtained variant list using ANNOVAR.
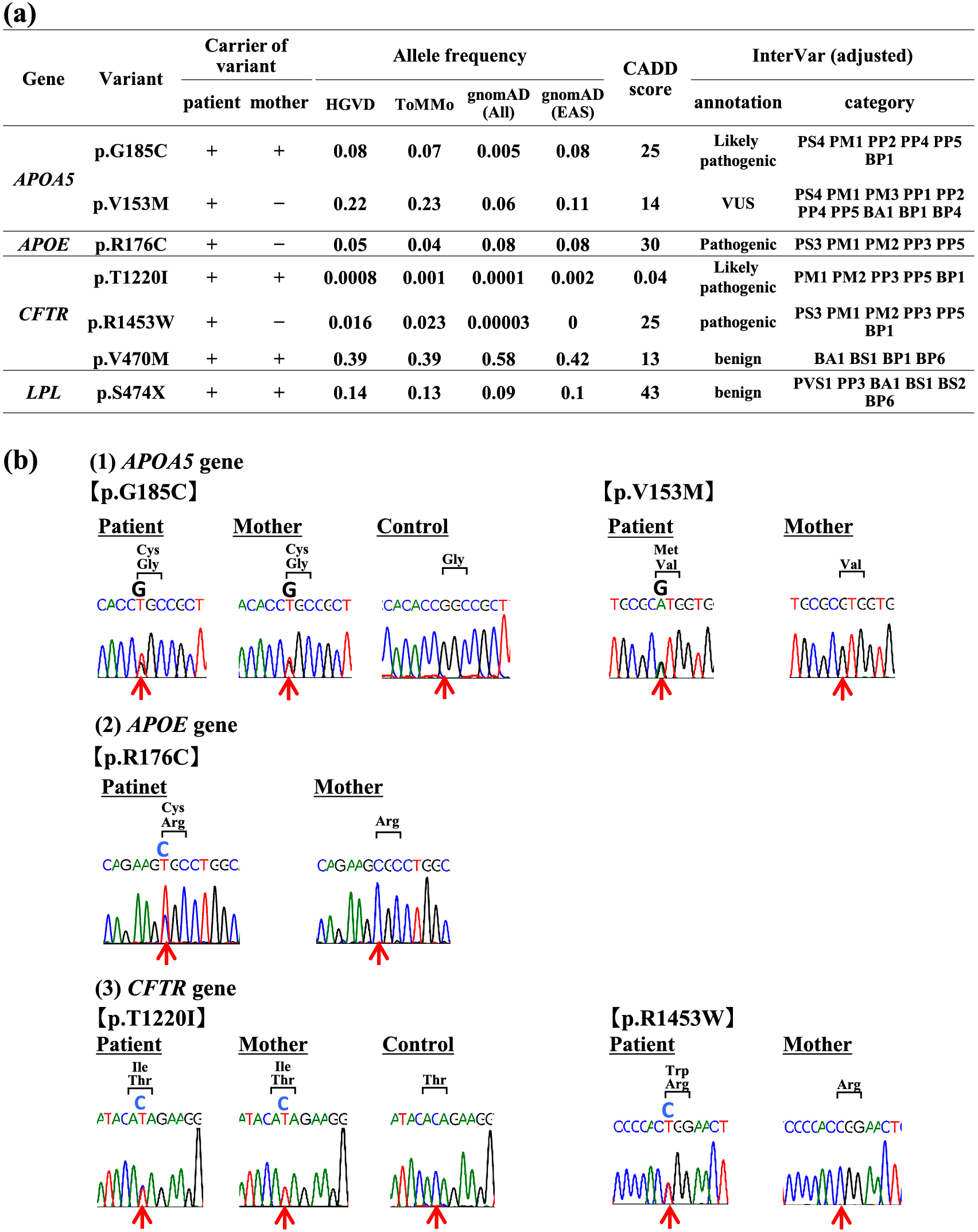
This study focused on variants of high pathological significance among six genes involved in hypertriglyceridemia (LPL, APOC2, GPIHBP1, LMF1, APOA5, APOE) [6, 7]. In addition, we examined variants among four genes involved in pancreatitis (Serine Protease 1 (PRSS1) for hereditary pancreatitis and Serine Peptidase Inhibitor Kazal Type 1 (SPINK1), Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), Carboxypeptidase A1 (CPA1) for idiopathic pancreatitis) [10, 11]. For the mutations found in these 10 candidate genes, we evaluated the pathogenicity of the mutations by using scoring the deleteriousness of single nucleotide variants by a combined annotation dependent depletion (CADD) (https://cadd.gs.washington.edu/) and minor allele frequency information in the HUMAN Genetic Variation Database (HGVD) (https://www.hgvd.genome.med.kyoto-u.ac.jp/), Tohoku Medical Megabank Organization databases (ToMMo) (https://jmorp.megabank.tohoku.ac.jp/202102/) and the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org) [12]. Furthermore, we obtained interpretation of pathogenicity by a bioinformatics tool InterVar. The pathogenicity of variants was adjusted using the guideline for interpretation of molecular sequencing by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) criteria [13]. The variants were classified into five classes: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign. As a result, we found that the patient had two heterozygous variants in the APOA5 gene; p.G185C (c.G553T, rs2075291) and p.V153M (c.G457A, rs3135507). Besides, we found that the patient had one heterozygous variant p.R176C (c.C526T, rs7412) in the APOE gene and p.S474X (c.C1421G, rs328) in the LPL gene. Furthermore, we found three heterozygous variants in the CFTR gene; p.T1220I (c.C3659T, rs1800123), p.R1453W (c.C4357T, rs4148725) and p.V470M (c.A1408G, rs213950) (Fig. 2a). In comparison, the mother had one heterozygous variant, p.G185C in the APOA5 gene and two heterozygous variants, p.T1220I and p.V470M in the CFTR gene (Fig. 2a). Her mother had the same LPL gene variant p.S474X. These variants were previously reported variants. Furthermore, confirmatory sanger sequencing was performed on the polymerase chain reaction (PCR) at five variants; p.G185C (APOA5 gene), p.V153M (APOA5 gene), p.R176C (APOE gene), p.T1220I (CFTR gene), and p.R1453W (CFTR gene) (Fig. 2b, Supplemental Table 1).
Discussion
WES analyses were used to evaluate the genes that cause hypertriglyceridemia and that are involved in the onset of acute pancreatitis. In the patient with severe hypertriglyceridemia and repeated acute pancreatitis, two heterozygous variants of the APOA5 gene (p.G185C and p.V153M), a heterozygous variant of the APOE gene (p.R176C), and three heterozygous variants of the CFTR gene (p.T1220I, p.R1453W, and p.V470M) were detected [14-18]. In contrast, a heterozygous variant in the APOA5 gene (p.G185C) and two heterozygous variants in the CFTR gene (p.T1220I and p.V470M) were detected in her mother, who had moderate hypertriglyceridemia without pancreatitis. The results suggested the more severe pathology of the patient than her mother might be due to the possible compound heterozygous variants in the APOA5 gene, the singular heterozygous variant in the APOE gene, and the possible compound heterozygous variants in the CFTR gene.
Within a short region on the chromosome 11q23-q24 is the human APOA5 gene, which is a member of the apolipoprotein gene family. Previously, ApoA5 was identified as a vital regulator of the serum TG concentration. The APOA5 gene is located on TG-rich lipoproteins (TGRLs), such as chylomicron and VLDL, and modulates LPL function by retaining TGRLs in the vascular endothelial cells involved in the progression of TGRL metabolism [19]. Most cases with a variant in the APOA5 gene exhibit hypertriglyceridemia in adulthood, characterized by an exacerbation of the condition under the influence of diabetes mellitus, obesity, and environmental factors, such as excessive alcohol consumption and carbohydrate and fat intake [20, 21]. A heterozygous variant—p.G185C—at exon 4 in the APOA5 gene has been reported as a causative gene for hypertriglyceridemia [14]. This same variant was found in both present cases, suggesting that p.G185C is probably involved in the pathogenesis of their hypertriglyceridemia. Another APOA5 gene variant—p.V153M—at exon 4, was found only in the patient, was not rare, and had a low CADD score of 14. According to the ACMG/AMP classification [13], p.V153M can be classified to PS1, PM1, PM3, PP1, PP2, PP4, PP5, BA1, BP1, and BP4 (Fig. 2a); thus, this variant is classified as a VUS (variant of uncertain significance) as per InterVar. However, Chen et al. reported that in a genetic analysis of eight genes (APOA5, APOE, Apolipoprotein C-III [APOC3], B Lymphoid Tyrosine Kinase (BLK), LPL, APOC2, GPIHBP1, and LMF1) associated with hypertriglyceridemia in eleven patients with TG >500 mg/dL and acute pancreatitis, one of the cases had only a p.V153M heterozygous variant in the APOA5 gene, suggesting that the variant may have caused hypertriglyceridemia [15]. In this study, genetic analyses for her mother as well as the patient revealed that the mother did not carry the p.V153M heterozygous variant observed in the patient, suggesting that this variant is possibly involved in the pathogenesis of severe hypertriglyceridemia in the patient. The mother retained a normal allele on one side of the APOA5 gene, while the patient might not possess any normal allele (compound heterozygous variants), leading to the difference in the severity of hypertriglyceridemia. However, it could also be possible that p.V153M was a de novo variant on the same allele as the p.G185C variant during the WES analysis for the patient and her mother.
ApoE is a type of apolipoprotein that is an essential component of chylomicrons, intermediate-density lipoproteins (IDLs), and VLDLs [16]. It is vital for the normal catabolism of triglyceride-rich lipoproteins. In the previously cited study by Chen et al., only one of the eleven cases had a p.R176C homozygous variant for the APOE gene, while another had a p.R176C heterozygous variant in the same gene and a p.V153M heterozygous variant in the APOA5 gene, suggesting that the p.R176C variant in the APOE gene is likely to be involved in the development of hypertriglyceridemia as well [15]. Presently, a p.R176C heterozygous variant in the APOE gene was found only in the patient. The p.R176C heterozygote was a low-frequency variant and had a CADD score as high as 30, suggesting that this variant may be one of the causes of hypertriglyceridemia in the patient. Additionally, in our study, a heterozygous variant—p.S474X—in the LPL gene was also detected. LPL is a representative enzyme of lipolysis [6]. It was found that the stop codon of the p.S474X heterozygous variant of the LPL gene was common in both the patient and her mother. Since the p.S474X was reported to increase LPL function [22], this variant seemed to neither cause hypertriglyceridemia in the patient nor her mother. Taken together, severe hypertriglyceridemia in the patients was considered to be affected not only by diabetes mellitus and obesity but also by these genetic factors such as possible compound heterozygous variants in the APOA5 gene (p.G185C and p.V153M) and the singular heterozygous variant in the APOE gene (p.R176C).
CFTR, encoded by the CFTR gene, is a significant anion channel in epithelial cells. Its dysfunction causes pancreatic fluid to become viscous and inflammatory, resulting in the development of pancreatitis [18]. The CFTR gene has been reported to be at an increased risk of dysfunction leading to pancreatitis if it carries compound heterozygous variants [23, 24]. In this case, the CFTR gene variant p.T1220I, found in both the patient and her mother, had a low CADD score of 0.04. However, according to ACMG/AMP classification [13], p.T1220I can be classified as PM1, PM2, PP3, PP5, and BP1 (Fig. 2a); thus, this variant is likely pathogenic as per InterVar. Furthermore, the variant was very rare frequency (MAF <0.001), suggesting that the variant is possibly pathogenic on the CFTR gene function. Whereas the p.T1220I heterozygous variant was found in both the patient and her mother, the p.R1453W heterozygous variant was found only in the patient. Additionally, the CADD score of p.R1453W was as high as 25. Lee et al. reported chloride (Cl–) channel activity of wild-type CFTR and R1453W using stably expressing Chinese hamster ovary (CHO)-K1 cells, and R1453W showed a decrease by 37% in whole-cell Cl– currents compared with the wild-type [25]. These results suggested that the p.R1453W heterozygous variant may reduce the CFTR gene function and may be involved in developing pancreatitis in our case. As two-thirds of the Japanese population carry the p.V470M variant since it was common with a low CADD score of 13 (Fig. 2a), it is considered to exhibit low pathogenicity.
Supplemental Fig. 2 summarizes the possible mechanism for acute pancreatitis. Based on our case report, it was suggested that severe hypertriglyceridemia could be affected by genetic predispositions, such as compound and singular heterozygous variants in the APOA5 and APOE genes, respectively, in addition to obesity and poor glycemic control. The precise mechanism(s) of severe hypertriglyceridemia inducing acute pancreatitis has not been fully elucidated. Hydrolysis of triglycerides with pancreatic lipase results in a high concentration of free fatty acids that provoke an inflammatory reaction in the pancreas [26]. Furthermore, hyperviscosity and ischemia could be caused by the accumulation of chylomicrons, which impairs microcirculation in the pancreas [27]. Moreover, susceptibility to pancreatitis, in this case, might also be increased by compound heterozygous variants in the CFTR gene [18, 25]. CFTR dysfunction was reported to affect pancreatic acinar cells using a CFTR (–/–) mouse model. This was done by impairing apical endocytosis through diminished ductal bicarbonate secretion and reduced alkalinization of the acinar lumen [28]. In this case, gallstones were detected on abdominal CT at the time of admission, but not on abdominal CT taken when the patient had pancreatitis in the past. However, it is undeniable that the stones may have been released from the common bile duct at the time of the CT imaging during pancreatitis. Since this is a case report, the generalization of this assessment should be made carefully. Further evidence is needed to shed light on this.
In this study, using WES analyses, we investigated the simultaneous genetic predisposition of individuals to hypertriglyceridemia and acute pancreatitis. Moreover, it became possible to consider the pathological significance of each gene variant detected by analyzing not only the patient but also her mother. As a result, we revealed that the patient has possible compound (p.G185C, p.V153M) and singular (p.R176C) heterozygous variants in the APOA5 and APOE genes, respectively. Furthermore, the patient possibly has compound heterozygous variants in the CFTR gene (p.T1220I, p.R1453W), which is a candidate for the onset of acute pancreatitis. This study indicates the clinical importance of WES analysis, which can simultaneously evaluate the genes that cause hypertriglyceridemia and that are involved in the onset of acute pancreatitis. Furthermore, the evaluation of genes involved in the latter condition may help identify high-risk cases that should be treated intensively among patients with hypertriglyceridemia.
Conflict of Interest
All authors declare no conflict of interest in this paper.
Disclosure Summary
The authors have nothing to disclose.
References
- 1 de Graaf J, Couture P, Sniderman A (2008) A diagnostic algorithm for the atherogenic apolipoprotein B dyslipoproteinemias. Nat Clin Pract Endocrinol Metab 4: 608–618.
- 2 Talmud PJ, Smart M, Presswood E, Cooper JA, Nicaud V, et al. (2008) ANGPTL4 E40K and T266M: effects on plasma triglyceride and hdl levels, postprandial responses, and CHD risk. Arterioscler Thromb Vasc Biol 28: 2319–2325.
- 3 Antonios N, Angiolillo DJ, Silliman S (2008) Hypertriglyceridemia and ischemic stroke. Eur Neurol 60: 269–278.
- 4 Murad MH, Hazem A, Coto-Yglesias F, Dzyubak S, Gupta S, et al. (2012) The association of hypertriglyceridemia with cardiovascular events and pancreatitis: a systematic review and meta-analysis. BMC Endocr Disord 12: 2.
- 5 Scherer J, Singh VP, Pitchumoni CS, Yadav D (2014) Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol 48: 195–203.
- 6 Brahm AJ, Hegele RA (2015) Chylomicronaemia—current diagnosis and future therapies. Nat Rev Endocrinol 11: 352–362.
- 7 Stroes E, Moulin P, Parhofer KG, Rebours V, Löhr JM, et al. (2017) Diagnostic algorithm for familial chylomicronemia syndrome. Atheroscler Suppl 23: 1–7.
- 8 Kobayashi J, Hashimoto H, Fukamachi I, Tashiro J, Shirai K, et al. (1993) Lipoprotein lipase mass and activity in severe hypertriglyceridemia. Clin Chim Acta 216: 113–123.
- 9 Murano T, Sako T, Oikawa S, Shirai K (2005) The recovery of dysfunctional lipoprotein lipase (Asp204-glu) activity by modification of substrate. Atherosclerosis 183: 101–107.
- 10 Zou WB, Tang XY, Zhou DZ, Qian YY, Hu LH, et al. (2018) Spink1, prss1, ctrc, and cftr genotypes influence disease onset and clinical outcomes in chronic pancreatitis. Clin Transl Gastroenterol 9: 204.
- 11 Masamune A (2014) Genetics of pancreatitis: the 2014 update. Tohoku J Exp Med 232: 69–77.
- 12 Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315.
- 13 Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424.
- 14 Pullinger CR, Aouizerat BE, Movsesyan I, Durlach V, Sijbrands EJ, et al. (2008) An apolipoprotein A-V gene SNP is associated with marked hypertriglyceridemia among Asian-American patients. J Lipid Res 49: 1846–1854.
- 15 Chen WJ, Sun XF, Zhang RX, Xu MJ, Dou TH, et al. (2017) Hypertriglyceridemic acute pancreatitis in emergency department: typical clinical features and genetic variants: HTGAP in emergency department. J Dig Dis 18: 359–368.
- 16 Mahley RW (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240: 622–630.
- 17 Xiao Y, Yuan W, Yu B, Guo Y, Xu X, et al. (2017) Targeted gene next-generation sequencing in chinese children with chronic pancreatitis and acute recurrent pancreatitis. J Pediatr 191: 158–163.e3.
- 18 Nakano E, Masamune A, Niihori T, Kume K, Hamada S, et al. (2015) Targeted next-generation sequencing effectively analyzed the cystic fibrosis transmembrane conductance regulator gene in pancreatitis. Dig Dis Sci 60: 1297–1307.
- 19 Talmud PJ (2007) Rare APOA5 mutations—clinical consequences, metabolic and functional effects: an ENID review. Atherosclerosis 194: 287–292.
- 20 Albers K, Schlein C, Wenner K, Lohse P, Bartelt A, et al. (2014) Homozygosity for a partial deletion of apoprotein A-V signal peptide results in intracellular missorting of the protein and chylomicronemia in a breast-fed infant. Atherosclerosis 233: 97–103.
- 21 Okazaki H, Goldstein JL, Brown MS, Liang G (2010) Lxr-srebp-1c-phospholipid transfer protein axis controls very low density lipoprotein (Vldl) particle size. J Biol Chem 285: 6801–6810.
- 22 Ranganathan G, Unal R, Pokrovskaya ID, Tripathi P, Rotter JI, et al. (2012) The lipoprotein lipase (Lpl) S447X gain of function variant involves increased mRNA translation. Atherosclerosis 221: 143–147.
- 23 Rosendahl J, Landt O, Bernadova J, Kovacs P, Teich N, et al. (2013) CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut 62: 582–592.
- 24 Noone PG, Zhou Z, Silverman LM, Jowell PS, Knowles MR, et al. (2001) Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology 121: 1310–1319.
- 25 Lee JH, Choi JH, Namkung W, Hanrahan JW, Chang J, et al. (2003) A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet 12: 2321–2332.
- 26 Havel RJ (1969) Pathogenesis, differentiation and management of hypertriglyceridemia. Adv Intern Med 15: 117–154.
- 27 Valdivielso P, Ramírez-Bueno A, Ewald N (2014) Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med 25: 689–694.
- 28 Freedman SD, Kern HF, Scheele GA (2001) Pancreatic acinar cell dysfunction in CFTR(–/–) mice is associated with impairments in luminal pH and endocytosis. Gastroenterology 121: 950–957.
