Abstract
Accumulating evidence has revealed that several conditions related to abnormal thyroid hormone status, such as dyslipidemia, hypertension, or hypercoagulable state, can exacerbate atherosclerotic vascular disease. Thyroid hormone effects on vascular smooth muscle cells and endothelial cells have also been studied extensively. However, only limited information is available on thyroid hormone-mediated immune response in current review articles on the pathophysiology of atherosclerosis. This report thus presents an overview of the recent advances in the understanding of the dynamic interactions taking place between thyroid hormone status and immune response in the pathogenesis of atherosclerosis. In particular, we focus on macrophages and T-lymphocytes, which have been recognized as important determinants for the initiation and development of atherosclerosis. Numerous studies have revealed the role of autophagy in immune cells produced in atherosclerosis. In addition, thyroid hormones induce autophagy in several cells and tissues, such as liver, skeletal muscles, lungs, and brown adipose tissue. Our research group, among others, have reported different targets of thyroid hormone-mediated autophagy, including lipid droplets (lipophagy), mitochondria (mitophagy), and aggregated proteins (aggrephagy). Based on these findings, thyroid hormone-mediated autophagy could serve as a novel therapeutic approach for atherosclerosis. We also consider the limitations of the current murine models for studies on atherosclerosis, especially in relation to low-density lipoprotein-cholesterol driven atherosclerotic plaque.
Introduction
Thyroid hormones regulate a wide range of systemic processes involved in metabolism and homeostasis [1, 2]. There is a considerable body of research on thyroid hormone status and atherosclerosis suggesting that both thyroid hormone deficiency and thyroid hormone excess can exacerbate atherosclerotic vascular disease [3].
Several conditions related to abnormal thyroid hormone status, such as dyslipidemia, hypertension, and hypercoagulable state, have been reported to predispose patients to atherosclerotic vascular disease [4-6]. In addition, thyroid hormone effects on vascular smooth muscle cells and endothelial cells, which are important for the initiation and development of atherosclerosis, have been extensively studied [4, 6]. However, in current review articles on atherosclerosis, limited information is available on the interactions between thyroid hormone status and immune response.
Autophagy is an essential intracellular degradation process that helps to maintain metabolic homeostasis by recycling the components of proteins and organelles [7]. Accumulated evidence has demonstrated that autophagy is associated with various diseases, such as malignant tumors and immune system disorders [8]. Autophagy has also been reported to play a pivotal role in regulating the immune response in atherosclerotic plaque [9]. For instance, the function of autophagy in macrophages has recently received increased attention. Additionally, a growing number of studies has provided evidence that thyroid hormones induce autophagy in various cells and tissues [10-13].
In this article, we review the literature and present an overview of recent advances in understanding the dynamic interactions between thyroid hormone status and immune response in the pathophysiological mechanisms of atherosclerosis. We also discuss the potential role of thyroid hormone-mediated autophagy as seen through the immune response in atherosclerosis.
Mechanisms of Thyroid Hormone Action
Circulating concentrations of thyroid hormones are tightly regulated by the hypothalamus–pituitary–thyroid axis in healthy individuals [14-16]. Briefly, thyrotropin-releasing hormone acts on the anterior pituitary gland to synthesize and secrete TSH, which then stimulates the thyroid gland to synthesize and release thyroid hormones, including the prohormone thyroxine (T4) and the active form triiodothyronine (T3).
It has been suggested that circulating thyroid hormone concentrations may not always reflect thyroid hormone action in specific peripheral tissues [17]. Cell culture and in vivo animal model studies revealed that thyroid hormone effects are not only determined by serum hormone concentrations but also by the expression levels of several key regulators, such as thyroid hormone transporters, iodothyronine deiodinases, thyroid hormone receptors (TRs), retinoid X receptors (RXRs), corepressors, and coactivators (Fig. 1) [18-20]. Circulating free thyroid hormones enter the cell via plasma membrane transporters, with type 2 iodothyronine deiodinases (D2) and type 3 iodothyronine deiodinases (D3), increasing and decreasing the intracellular concentrations of the T3 active form, respectively. T3 molecules enter the nucleus, where they bind to the nuclear TRs mainly as heterodimers with RXRs. TRs are encoded by thyroid hormone receptor alpha (THRA or TRα) and thyroid hormone receptor beta (THRB or TRβ) genes [18]. TRα1 and TRα2 mRNAs are generated via alternative splicing of TRα, whereas the use of different promoters in TRβ generates TRβ1 and TRβ2 mRNAs. In T3-dependent initiation of transcription, ligand-bound TRs bind to thyroid hormone response elements, which are typically located in the promoter regions of target genes, and recruit coactivator complexes that transactivate T3-regulated target genes.
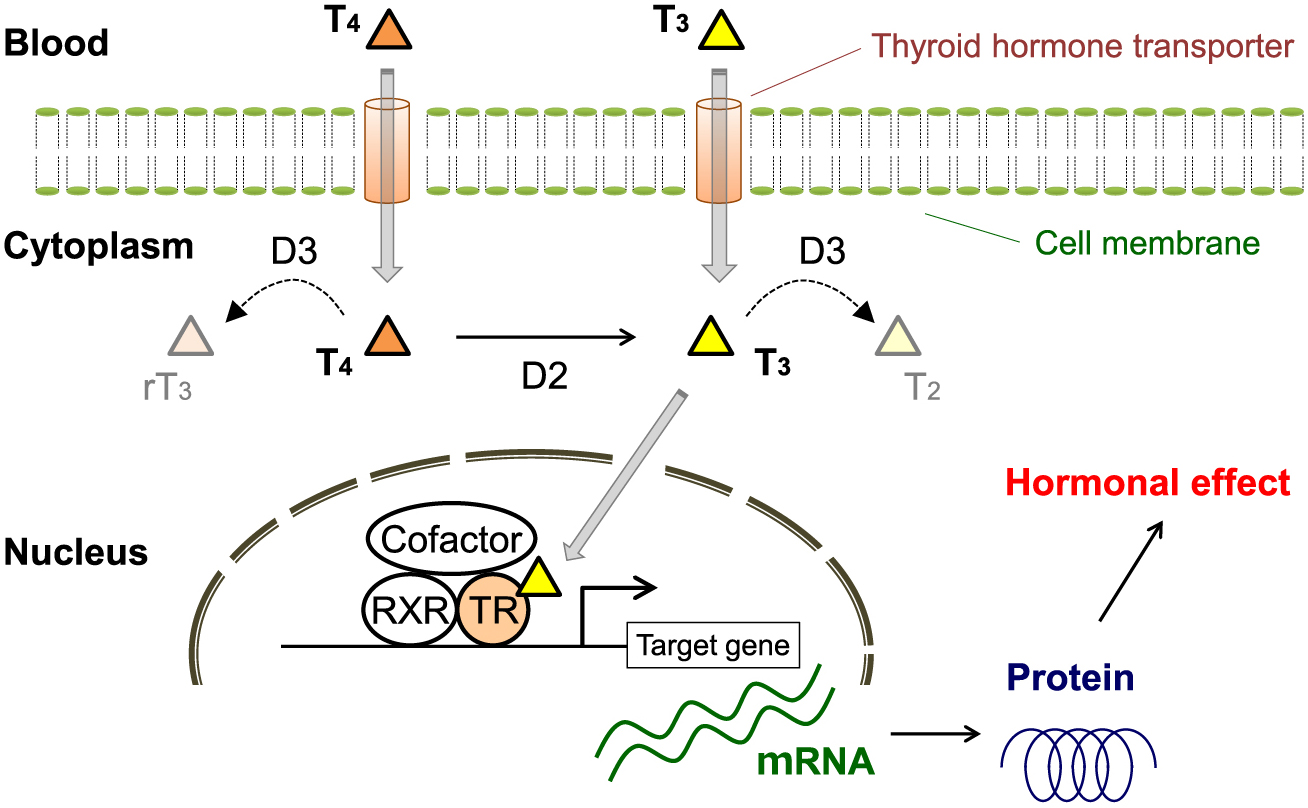
Thyroid Hormone Status and Atherosclerotic Risk Factors
The accelerated process of atherosclerosis associated with thyroid dysfunction has been traditionally ascribed to several conditions, including dyslipidemia, hypertension, impaired endothelial function, and hypercoagulable state [4-6]. Before reviewing the potential mediating role of the immune response in atherosclerosis, this section provides a brief overview of the thyroid hormone effects on the risk factors for atherosclerosis.
First, thyroid hormones regulate lipid metabolism by multiple mechanisms. For example, thyroid hormones enhance the expression of hepatic low-density lipoprotein receptor (Ldlr) to increase serum cholesterol clearance [21]. Recent reviews describe the details of the thyroid hormone status and atherosclerotic lipid profiles [22]. Second, thyroid hormones play an essential role in the regulation of blood pressure. Sufficient evidence has revealed that both thyroid hormone deficiency and thyroid hormone excess increase the blood pressure [4, 6]. Third, thyroid hormones regulate endothelial nitric oxide production and the vascular tone [4, 6]. It has been also reported that patients with hypothyroidism or hyperthyroidism exhibit impaired endothelial function, which is known to be one of the earliest steps in the development of atherosclerosis. Finally, detailed information on thyroid hormone effects on other risk factors, such as a hypercoagulable state and hyperhomocysteinemia, has been discussed elsewhere [4-6].
Thyroid Hormone Status and Immune Response in Atherosclerosis
Thyroid hormones exert pleiotropic effects on immune response. Previous studies have identified thyroid hormone effects on the functions of both innate immune cells (e.g., granulocytes, natural killer cells, macrophages, and dendritic cells) and adaptive immune cells (e.g., T- and B-lymphocytes) [23]. Here, we focus on macrophages and T-lymphocytes, which have been recognized as important determinants in the pathogenesis of atherosclerosis [24, 25].
Atherosclerotic lesion formation falls under four categories: fatty streak lesions, established lesions, vulnerable plaques, and rupture-prone plaques [24]. Macrophages play an important role in all stages of atherosclerosis. To our knowledge, there are only two studies investigating thyroid hormone effects on macrophage function using rodent models of atherosclerosis. Mörk et al. found that the thyromimetic compound KB3495 exerts a cholesterol-independent positive effect on atherosclerotic plaque formation in apolipoprotein E (apoE)-deficient mice [26] which serve as a classic model for studying atherosclerosis. Treatment with KB3495 resulted in reduced macrophage content in atherosclerotic plaques as well as a significant reduction in serum levels of inflammatory cytokines (e.g., interleukin-1β (IL-1β), tumor necrosis factor-α, and interleukin-6). Billon et al. found that additional deletion of TRα resulted in impaired macrophage cholesterol efflux and earlier formation of atherosclerotic plaques in apoE-deficient mice [27]. The same group also demonstrated that the absence of TRα was associated with increased macrophage-mediated inflammation, without affecting lipid profiles or blood pressure. Furuya et al. utilized a mouse model of chronic kidney disease and reported that a lack of TRα results in increased macrophage-mediated inflammation via the nuclear factor-κB pathway [28]. These findings suggest that increased T3 action shifts macrophages towards an anti-inflammatory status, whereas decreased signaling through ligand-bound TRα results in a proinflammatory status.
In addition to TRα, TRβ is expressed in macrophages [29]. Although TRβ plays a critical role in mediating T3 effects on murine bone marrow-derived macrophages (BMDMs) [29], the relevance of TRβ in atherosclerosis remains unclear. In addition to these TRs, macrophages also express deiodinases and thyroid hormone transporters. Several studies have evaluated the effects of these regulators on inflammatory responses. For example, previous studies using BMDMs extracted from D2 knockout mice have suggested that decreased intracellular T3 concentrations influence macrophage polarization and have an anti-inflammatory effect against lipopolysaccharide-induced responses [30, 31]. However, to our knowledge, there have been no studies exploring the effects of these regulators on macrophage function in atherosclerosis. Further studies on the molecular pathways of thyroid hormone metabolism in macrophages could identify potential pharmacological targets in patients with atherosclerotic vascular disease.
In addition to macrophages, T-lymphocytes have been shown to play pivotal roles in atherosclerosis [25]. T-cell subsets are categorized into two major subtypes, CD4+ T-lymphocytes and CD8+ T-lymphocytes, based on their cellular functions and capacity to secrete specific cytokines. These subsets are further classified into effector T-lymphocytes (Teffs) and regulatory T-lymphocytes (Tregs). Recent experimental evidence has revealed that Teffs promote atherosclerotic disorders, whereas Tregs inhibit atherosclerotic progression by dampening Teff-mediated responses [32].
Regarding the association between thyroid hormone status and T-lymphocytes, Mihara et al. demonstrated that human peripheral CD4+ T-lymphocytes cultured with T3 showed enhanced apoptosis compared to the apoptosis in those cultured without T3 [33]. The same group suggested that thyroid hormones may reduce Teff-mediated atherosclerogenic responses by inducing apoptosis. On the other hand, Zhong et al. evaluated blood samples from 30 patients with Graves’ hyperthyroidism and found that high circulating levels of thyroid hormones impaired Treg function via reduced programmed death 1 (PD-1) expression [34]. Tregs have been shown to play a protective role against atherosclerosis, hence excess amounts of thyroid hormones may be harmful for atherosclerotic disorders. Overall, these findings suggest that optimal intracellular T3 concentrations are essential for the anti-atherosclerogenic function of T-lymphocytes. However, further studies are required to evaluate the possible role of T-lymphocytes using in vitro and in vivo models of atherosclerosis.
TSH has been identified to exert organ-specific modulating effects by releasing thyroid hormones from thyrocytes that express the TSH receptor (TSHR). In addition to thyrocytes, TSHR has been reported to be expressed in a variety of cells, such as osteoclasts [35] and hepatocytes [36]. Several studies suggest that TSH can exert cell type-specific modulating effects not only by stimulating the thyroid gland to secrete thyroid hormones but also by directly affecting these cells through downstream pathways of TSH-bound TSHR. For example, Tian et al. demonstrated that ligand-bound TSHR in hepatocytes modulated liver cholesterol metabolism [36]. Importantly, TSHR expression has also been identified in macrophages [37]. A recent study using apoE-deficient mice demonstrated that TSH can contribute to atherosclerosis by promoting macrophage inflammation in atherosclerotic plaques [38]. The same research group further revealed that TSH activates macrophage-mediated inflammation via G-protein mediated pathways, such as the Gα13-p38 and Gα15-PLC-IκB pathways [39]. Collectively, these findings support the notion that TSH can regulate macrophage functions, while in vivo studies confirm that the direct action of TSH could be difficult to interpret because of the concomitant changes in the serum concentrations of thyroid hormones.
TSHRs were also reported to be expressed in T-lymphocytes [40], but their functions remain ambiguous. The possible direct effect of TSH on T-lymphocytes is also an interesting issue that needs to be investigated further.
Autophagy and Thyroid Hormone Status
Autophagy is classified into three categories: macroautophagy, chaperone-mediated autophagy, and microautophagy [7, 8]. Macroautophagy, the most extensively studied type of autophagic processes, mainly involves the following steps. After cup-shaped structures called phagophores are generated, the subsequent elongation and expansion of phagophores form complete double-membrane sphere vesicles called autophagosomes. The autophagosomes then fuse with lysosomes and transform to autolysosomes, where proteins and organelles are digested for recycling. Macroautophagy (hereafter referred to as autophagy) was initially described as a non-selective bulk process induced by starvation. In addition to non-selective autophagy, selective autophagy has been clarified as a tightly regulated process that requires cargo (target) recognition.
Although thyroid hormone-mediated activation of lysosomal enzymes was first reported in the late 1960s, the possible role of autophagy in the pathogenesis of thyroid diseases was not clarified for a long time [7]. Since thyroid hormone-induced autophagy was first reported by Sinha et al. in 2012 [41], numerous studies have demonstrated the close correlation between thyroid disorders and autophagy. Major topics include thyroid hormone effects on autophagic signaling (thyroid hormone-mediated autophagy) [10-13], the relationship between autophagy and the development of thyroid diseases (e.g., thyroid carcinoma or autoimmune thyroid diseases) [42], and the role of autophagy in the quality control of thyrocytes [43]. In line with the first topic, our research group, among others, have identified different cargos (targets) of thyroid hormone-mediated autophagy, including lipid droplets (lipophagy), mitochondria (mitophagy), and aggregated proteins (aggrephagy) [2, 12, 44, 45].
The term lipophagy was proposed by Singh et al. to emphasize the role of autophagy in lipid metabolism [46]. This process involves selective delivery of lipid droplets for lysosomal degradation. Sinha et al. revealed that thyroid hormones induce lipophagy in both murine and human hepatic cells [41]. Another group further revealed a pivotal role of hepatic chromosome 19 open reading frame 80 (C19orf80; also known as angiopoietin-like protein 8 (ANGPTL8)) in thyroid hormone-mediated lipophagy [44]. More recently, Zhou et al. have demonstrated that mediator complex subunit 1 (MED1) is a key participant in promoting autophagy and lipophagy in the liver during different nutrient and hormonal conditions (Fig. 2) [47].
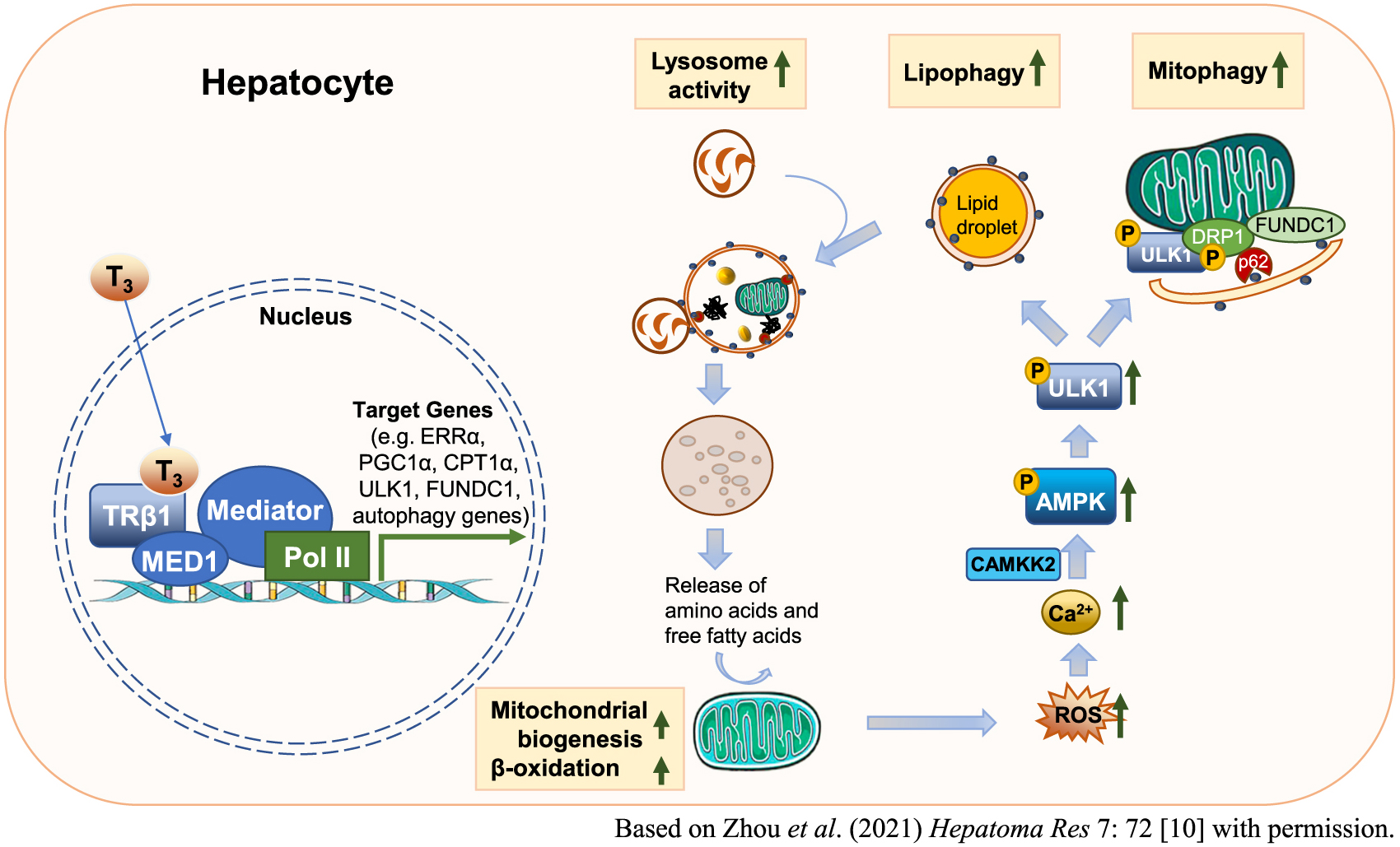
Mitochondrial quality is maintained by various mechanisms including fission, fusion, and autophagy. In this connection, the term mitophagy was proposed by Lemasters to indicate the process of selective removal of mitochondria by autophagy [48]. Sinha et al. reported that ligand-bound TR induces mitophagy to reduce cellular injury caused by reactive oxygen species (ROS) in hepatic cells (Fig. 2) [49]. The same group further reported that estrogen-related receptor alpha (ERRα), an orphan nuclear receptor, coordinately regulates mitophagy along with TR in the liver [50]. Besides lipophagy and mitophagy, thyroid hormone-mediated autophagy has also been linked to increased cellular survival by preventing protein aggregate formation in the liver [45].
An essential role of thyroid hormone-mediated autophagy has been reported not only in the liver but also in several other organs [11-13]. For example, Lesmana et al. demonstrated that thyroid hormone-mediated autophagy plays a significant role in stimulating mitochondrial activity and biogenesis in skeletal muscles [11]. The same group further clarified the importance of TRα1 signaling for autophagy, mitochondrial biogenesis, and fatty acid use in skeletal muscles [51]. These findings suggest that a deeper understanding of hormonal or pharmacological induction of TR and other nuclear receptors related to autophagic signaling may lead to the development of new therapeutic strategies for metabolic disorders and aging.
Autophagy and Immune Response in Atherosclerosis
Accumulated evidence has revealed that autophagy occurs during atherosclerosis [9]. Transmission electron microscopy has been used to confirm the formation of autophagosomes in primary cell types of human atherosclerotic plaque, including macrophages, vascular smooth muscle cells, and endothelial cells [52]. This section discusses the role of autophagy in immune cells (i.e., macrophages and T-lymphocytes) against atherosclerosis.
The protective effects of autophagy in macrophages include reducing apoptosis, deactivating inflammasomes, and facilitating reverse cholesterol transport (RCT) [53]. In experiments using fat-fed Ldlr-deficient mice, a mouse model of advanced atherosclerosis, Liao et al. demonstrated that the knockdown of autophagic gene ATG5 renders macrophages more susceptible to apoptosis, worsens the recognition and clearance of dead cells (efferocytosis), and promotes plaque necrosis [53]. Inflammasomes are multimeric protein complexes that regulate the activation of caspase-1 and induce inflammation in response to harmful stimuli, such as infectious microbes or molecules derived from host proteins as reported by Guo et al. [54]. Activated caspase-1 cleaves proinflammatory cytokines, including IL-1β. Razani et al. demonstrated that the knockdown of ATG5 in macrophages promotes atherosclerosis, in part through inflammasome hyperactivation in apoE-deficient mice [55]. The same group also proposed that disturbed mitophagy is a possible mechanism for inflammasome activation in macrophages [55].
Selective autophagy of lipid droplets, lipophagy, has also been identified to play an essential role in macrophages against atherosclerosis [56, 57]. Lipid droplets are the major sites of cholesterol storage in macrophage foam cells and are a key component of atherosclerotic lesions. The process of cholesterol efflux from the peripheral tissues to the liver is referred to as RCT. Using murine BMDMs, Ouimet et al. provided evidence that cytosolic lipid droplets are delivered to lysosomes via lipophagy [56]. The same group also reported that lysosomal acid lipase (LAL) hydrolyzes cholesterol esters in lipid droplets to generate free cholesterol, mainly for ATP-binding cassette transporter 1 (ABCA1)-dependent efflux. Another study by Le Guezennec et al. identified a role for Wip1 phosphatase, a negative regulator of Atm-dependent signaling, in macrophage RCT using apoE-deficient mice [57]. The same group demonstrated that Wip1-dependent regulation is important for the conversion of macrophages into foam cells via lipophagy. More recently, Zhang et al. have demonstrated that an adipokine, C1q/tumor necrosis factor-related protein-9 (CTRP9), protects macrophages against atherosclerosis by enhancing autophagy to promote cholesterol efflux [58]. These findings suggest that lipophagy is an important factor in macrophage RCT.
Several inducers of autophagy have been studied to target autophagic signaling for the treatment of atherosclerosis. First, the selective inhibition of the phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt)-mammalian target of rapamycin (mTOR) pathway has been shown to stabilize vulnerable atherosclerotic plaques by promoting macrophage autophagy [59]. There is evidence that mTOR inhibitors promote plaque stability in animal models and clinical studies [60, 61]. Second, Liu et al. revealed that the application of Sirtuin1 (Sirt1) activator, resveratrol, increased the efferocytosis of oxidized low-density lipoprotein (LDL)-induced apoptotic cells through the upregulation of autophagy [62]. The same group also proposed Sirt1 as a novel therapeutic target for the treatment of atherosclerosis. Given that thyroid hormones induce autophagy in several cells and tissues [10-13], we deduced that thyroid hormones can promote macrophage autophagy and prevent the progression of atherosclerosis (Fig. 3). Additionally, we revealed that thyroid hormones regulate the transcription of a subset of hepatic target genes by Sirt1-mTOR complex 2 (mTORC2)-Akt signaling [63]. Hence the thyroid hormone-mediated activation of Sirt1-signaling could also serve as a novel therapeutic approach for atherosclerosis. However, the deleterious effects of autophagy have also been reported in certain human disease settings [64]. For instance, excessive autophagy results in autophagy-dependent cell death termed autosis, which can contribute to plaque destabilization [9]. Further studies are necessary to identify the protective and detrimental effects of autophagy on macrophages in relation to atherosclerosis.

An imbalance in the proportion of Teffs and Tregs is associated with the progression of atherosclerosis [32]. In this context, autophagy has been shown to be essential for the cell-intrinsic process required to maintain the functional integrity of Tregs [65]. The possible effect of thyroid hormone-mediated autophagy on T-lymphocytes is not yet well understood, hence further investigation would be of great interest.
Limitations of Current Mouse Models for the Study of Atherosclerosis
The pathogenesis of atherosclerosis has been extensively studied in mice, but there is only limited information available on the LDL-cholesterol-dominant model. Dyslipidemia is classified into six categories according to elevated lipoprotein levels as follows: type I (high chylomicrons; CM), type IIa (high LDL), type IIb (high LDL and very low-density lipoprotein; VLDL), type III (high intermediate-density lipoprotein; IDL), type IV (high VLDL), and type V (high CM and VLDL) [66]. The frequencies of each type in Japanese male population were reported as 0.1%, 32.2%, 20.9%, 0.3%, 44.6%, and 1.9%, respectively; while those in Japanese female population were reported as 0.3%, 53.3%, 22.8%, 0.7%, 21.5%, and 1.4%, respectively [67]. Although mice deficient in apoE or Ldlr have been extensively used, mice that are fed a normal chow diet do not mimic type IIa dyslipidemia, which is one of the most common types in human dyslipidemia [68-70].
To resolve the limited utility of apoE or Lldr-deficient mice, Powell-Braxton et al. generated a mouse model with double deficiency of Ldlr and apolipoprotein B editing enzyme 1 (Apobec 1; hereafter referred to as L–/–/A–/– mice). These L–/–/A–/– mice exhibited a marked elevation in apolipoprotein B-100 containing LDL, closely mirroring the plasma lipid profiles of human type IIa dyslipidemia, and developed extensive atherosclerosis even on a low-fat chow diet [71]. We have recently revealed that the L–/–/A–/– mice develop atherosclerotic plaques with several key features of LDL-driven atherosclerosis reported in the patients with type IIa dyslipidemia [69]. We have also provided evidence that this mouse model is useful for studying the pathophysiology of several diseases, including fibrinogen deficiency, plasminogen deficiency, and aortic aneurysms, in relation to LDL-cholesterol-driven atherosclerosis [68, 70, 72].
As per the available literature, studies that use L–/–/A–/– mice for determining the importance of thyroid hormones in the pathogenesis of atherosclerosis are lacking. The potential role of thyroid hormone-mediated immune response and autophagy in LDL-cholesterol-dominant mouse models are thus important issues that need to be addressed in future studies.
Conclusions
In conclusion, accumulated evidence has revealed that thyroid hormone status affects the immune response through several possible mechanisms in the pathophysiology of atherosclerosis. Additionally, the protective role of autophagy in the immune response against atherosclerosis has been extensively investigated. Since thyroid hormones have been shown to induce autophagy in several cells and tissues (Fig. 2), they can also induce autophagic signaling in immune cells to prevent the progression of atherosclerosis (Fig. 3). A deeper understanding of thyroid hormone-mediated autophagy may lead to the development of new therapeutic strategies for patients with atherosclerotic vascular diseases.
Financial Support
This work was supported in part by KAKENHI Grants (19K17981 for KO, 21K08772 for TI) from the Japan Society for the Promotion of Science.
Disclosure
The authors have nothing to declare.
References
- 1 Yen PM (2001) Physiological and molecular basis of thyroid hormone action. Physiol Rev 81: 1097–1142.
- 2 Singh BK, Sinha RA, Ohba K, Yen PM (2017) Role of thyroid hormone in hepatic gene regulation, chromatin remodeling, and autophagy. Mol Cell Endocrinol 458: 160–168.
- 3 Cappola AR, Desai AS, Medici M, Cooper LS, Egan D, et al. (2019) Thyroid and cardiovascular disease: research agenda for enhancing knowledge, prevention, and treatment. Thyroid 29: 760–777.
- 4 Ichiki T (2016) Thyroid hormone and vascular remodeling. J Atheroscler Thromb 23: 266–275.
- 5 Ellervik C, Mora S, Kuś A, Åsvold B, Marouli E, et al. (2021) Effects of thyroid function on hemostasis, coagulation, and fibrinolysis: a mendelian randomization study. Thyroid 31: 1305–1315.
- 6 Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, et al. (2018) Thyroid hormones and cardiovascular function and diseases. J Am Coll Cardiol 71: 1781–1796.
- 7 Sinha RA, Singh BK, Yen PM (2017) Reciprocal crosstalk between autophagic and endocrine signaling in metabolic homeostasis. Endocr Rev 38: 69–102.
- 8 Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741.
- 9 Xu J, Kitada M, Ogura Y, Koya D (2021) Relationship between autophagy and metabolic syndrome characteristics in the pathogenesis of atherosclerosis. Front Cell Dev Biol 9: 641852.
- 10 Zhou J, Sinha RA, Yen PM (2021) The roles of autophagy and thyroid hormone in the pathogenesis and treatment of NAFLD. Hepatoma Res 7: 72.
- 11 Lesmana R, Sinha RA, Singh BK, Zhou J, Ohba K, et al. (2016) Thyroid hormone stimulation of autophagy is essential for mitochondrial biogenesis and activity in skeletal muscle. Endocrinology 157: 23–38.
- 12 Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, et al. (2018) Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med 24: 39–49.
- 13 Yau WW, Singh BK, Lesmana R, Zhou J, Sinha RA, et al. (2019) Thyroid hormone (T(3)) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 15: 131–150.
- 14 Hollenberg AN (2013) Regulation of thyrotropin secretion. In: Braverman LE, Cooper DS (eds) Werner & Ingbar’s the thyroid: a fundamental and clinical text (10th). Lippincott Williams & Wilkins, Philadelphia, USA: 169–182.
- 15 Ohba K (2021) An update on the pathophysiology and diagnosis of inappropriate secretion of thyroid-stimulating hormone. Int J Mol Sci 22: 6611.
- 16 Sakai Y, Ohba K, Sasaki S, Matsushita A, Nakamura HM, et al. (2021) Impairment of the hypothalamus-pituitary-thyroid axis caused by naturally occurring GATA2 mutations in vitro. Int J Mol Sci 22: 10015.
- 17 Ohba K, Leow MK, Singh BK, Sinha RA, Lesmana R, et al. (2016) Desensitization and incomplete recovery of hepatic target genes after chronic thyroid hormone treatment and withdrawal in male adult mice. Endocrinology 157: 1660–1672.
- 18 Cheng SY, Leonard JL, Davis PJ (2010) Molecular aspects of thyroid hormone actions. Endocr Rev 31: 139–170.
- 19 Brent GA (2012) Mechanisms of thyroid hormone action. J Clin Invest 122: 3035–3043.
- 20 Ohba K, Sinha RA, Singh BK, Iannucci LF, Zhou J, et al. (2017) Changes in hepatic TRβ protein expression, lipogenic gene expression, and long-chain acylcarnitine levels during chronic hyperthyroidism and triiodothyronine withdrawal in a mouse model. Thyroid 27: 852–860.
- 21 Lopez D, Abisambra Socarras JF, Bedi M, Ness GC (2007) Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim Biophys Acta 1771: 1216–1225.
- 22 Ritter MJ, Amano I, Hollenberg AN (2020) Thyroid hormone signaling and the liver. Hepatology 72: 742–752.
- 23 Klein JR (2021) Dynamic interactions between the immune system and the neuroendocrine system in health and disease. Front Endocrinol (Lausanne) 12: 655982.
- 24 Moore KJ, Tabas I (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355.
- 25 Ait-Oufella H, Lavillegrand JR, Tedgui A (2021) Regulatory T cell-enhancing therapies to treat atherosclerosis. Cells 10: 723.
- 26 Mörk LM, Rehnmark S, Davoodpour P, Norata GD, Larsson L, et al. (2013) The thyroid receptor modulator KB3495 reduces atherosclerosis independently of total cholesterol in the circulation in ApoE deficient mice. PloS One 8: e78534.
- 27 Billon C, Canaple L, Fleury S, Deloire A, Beylot M, et al. (2014) TRα protects against atherosclerosis in male mice: identification of a novel anti-inflammatory property for TRα in mice. Endocrinology 155: 2735–2745.
- 28 Furuya F, Ishii T, Tamura S, Takahashi K, Kobayashi H, et al. (2017) The ligand-bound thyroid hormone receptor in macrophages ameliorates kidney injury via inhibition of nuclear factor-κB activities. Sci Rep 7: 43960.
- 29 Perrotta C, Buldorini M, Assi E, Cazzato D, De Palma C, et al. (2014) The thyroid hormone triiodothyronine controls macrophage maturation and functions: protective role during inflammation. Am J Pathol 184: 230–247.
- 30 Kwakkel J, Surovtseva OV, de Vries EM, Stap J, Fliers E, et al. (2014) A novel role for the thyroid hormone-activating enzyme type 2 deiodinase in the inflammatory response of macrophages. Endocrinology 155: 2725–2734.
- 31 van der Spek AH, Surovtseva OV, Jim KK, van Oudenaren A, Brouwer MC, et al. (2018) Regulation of intracellular triiodothyronine is essential for optimal macrophage function. Endocrinology 159: 2241–2252.
- 32 Tanaka T, Sasaki N, Rikitake Y (2021) Recent advances on the role and therapeutic potential of regulatory T cells in atherosclerosis. J Clin Med 10: 5907.
- 33 Mihara S, Suzuki N, Wakisaka S, Suzuki S, Sekita N, et al. (1999) Effects of thyroid hormones on apoptotic cell death of human lymphocytes. J Clin Endocrinol Metab 84: 1378–1385.
- 34 Zhong Y, Lu TT, Liu XM, Liu BL, Hu Y, et al. (2021) High levels of thyroid hormone impair regulatory T cell function via reduced PD-1 expression. J Clin Endocrinol Metab 106: 2738–2753.
- 35 Abe E, Marians RC, Yu W, Wu XB, Ando T, et al. (2003) TSH is a negative regulator of skeletal remodeling. Cell 115: 151–162.
- 36 Tian L, Song Y, Xing M, Zhang W, Ning G, et al. (2010) A novel role for thyroid-stimulating hormone: up-regulation of hepatic 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase expression through the cyclic adenosine monophosphate/protein kinase A/cyclic adenosine monophosphate-responsive element binding protein pathway. Hepatology 52: 1401–1409.
- 37 Coutelier JP, Kehrl JH, Bellur SS, Kohn LD, Notkins AL, et al. (1990) Binding and functional effects of thyroid stimulating hormone on human immune cells. J Clin Immunol 10: 204–210.
- 38 Yang C, Lu M, Chen W, He Z, Hou X, et al. (2019) Thyrotropin aggravates atherosclerosis by promoting macrophage inflammation in plaques. J Exp Med 216: 1182–1198.
- 39 Yang C, He Z, Zhang Q, Lu M, Zhao J, et al. (2021) TSH activates macrophage inflammation by G13- and G15-dependent Pathways. Endocrinology 162: bqab077.
- 40 Bağriaçik EU, Klein JR (2000) The thyrotropin (thyroid-stimulating hormone) receptor is expressed on murine dendritic cells and on a subset of CD45RBhigh lymph node T cells: functional role for thyroid-stimulating hormone during immune activation. J Immunol 164: 6158–6165.
- 41 Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, et al. (2012) Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest 122: 2428–2438.
- 42 Wei W, Hardin H, Luo QY (2019) Targeting autophagy in thyroid cancers. Endocr Relat Cancer 26: R181–R194.
- 43 Kurashige T, Nakajima Y, Shimamura M, Matsuyama M, Yamada M, et al. (2019) Basal autophagy deficiency causes thyroid follicular epithelial cell death in mice. Endocrinology 160: 2085–2092.
- 44 Tseng YH, Ke PY, Liao CJ, Wu SM, Chi HC, et al. (2014) Chromosome 19 open reading frame 80 is upregulated by thyroid hormone and modulates autophagy and lipid metabolism. Autophagy 10: 20–31.
- 45 Chi HC, Chen SL, Tsai CY, Chuang WY, Huang YH, et al. (2016) Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy 12: 2271–2285.
- 46 Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. (2009) Autophagy regulates lipid metabolism. Nature 458: 1131–1135.
- 47 Zhou J, Singh BK, Ho JP, Lim A, Bruinstroop E, et al. (2021) MED1 mediator subunit is a key regulator of hepatic autophagy and lipid metabolism. Autophagy 17: 4043–4061.
- 48 Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8: 3–5.
- 49 Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, et al. (2015) Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 11: 1341–1357.
- 50 Singh BK, Sinha RA, Tripathi M, Mendoza A, Ohba K, et al. (2018) Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci Signal 11: eaam5855.
- 51 Zhou J, Gauthier K, Ho JP, Lim A, Zhu XG, et al. (2021) Thyroid hormone receptor α regulates autophagy, mitochondrial biogenesis, and fatty acid use in skeletal muscle. Endocrinology 162: bqab112.
- 52 Perrotta I (2013) The use of electron microscopy for the detection of autophagy in human atherosclerosis. Micron 50: 7–13.
- 53 Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, et al. (2012) Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15: 545–553.
- 54 Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687.
- 55 Razani B, Feng C, Coleman T, Emanuel R, Wen H, et al. (2012) Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15: 534–544.
- 56 Ouimet M, Franklin V, Mak E, Liao X, Tabas I, et al. (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13: 655–667.
- 57 Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, et al. (2012) Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab 16: 68–80.
- 58 Zhang L, Liu Q, Zhang H, Wang XD, Chen SY, et al. (2018) C1q/TNF-related protein 9 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. J Cardiovasc Pharmacol 72: 167–175.
- 59 Zhai C, Cheng J, Mujahid H, Wang H, Kong J, et al. (2014) Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PloS One 9: e90563.
- 60 Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, et al. (2007) Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol 49: 706–715.
- 61 Jinnouchi H, Guo L, Sakamoto A, Sato Y, Cornelissen A, et al. (2020) Advances in mammalian target of rapamycin kinase inhibitors: application to devices used in the treatment of coronary artery disease. Future Med Chem 12: 1181–1195.
- 62 Liu B, Zhang B, Guo R, Li S, Xu Y (2014) Enhancement in efferocytosis of oxidized low-density lipoprotein-induced apoptotic RAW264.7 cells through Sirt1-mediated autophagy. Int J Mol Med 33: 523–533.
- 63 Singh BK, Sinha RA, Zhou J, Tripathi M, Ohba K, et al. (2016) Hepatic FOXO1 target genes are co-regulated by thyroid hormone via RICTOR protein deacetylation and MTORC2-AKT protein inhibition. J Biol Chem 291: 198–214.
- 64 Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22: 367–376.
- 65 Wei J, Long L, Yang K, Guy C, Shrestha S, et al. (2016) Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 17: 277–285.
- 66 Fredrickson DS (1971) An international classification of hyperlipidemias and hyperlipoproteinemias. Ann Intern Med 75: 471–472.
- 67 Matsuzawa Y, Tarui S (1989) Recent trend in the research of hyperlipidemia in Japan (1). Profiles of primary hyperlipidemia. Nihon Naika Gakkai Zasshi 78: 1396–1369 (in Japanese).
- 68 Iwaki T, Sandoval-Cooper MJ, Brechmann M, Ploplis VA, Castellino FJ (2006) A fibrinogen deficiency accelerates the initiation of LDL cholesterol-driven atherosclerosis via thrombin generation and platelet activation in genetically predisposed mice. Blood 107: 3883–3891.
- 69 Miyajima C, Iwaki T, Umemura K, Ploplis VA, Castellino FJ (2018) Characterization of atherosclerosis formation in a murine model of type IIa human familial hypercholesterolemia. Biomed Res Int 2018: 1878964.
- 70 Iwaki T, Arakawa T, Sandoval-Cooper MJ, Smith DL, Donahue D, et al. (2021) Plasminogen deficiency significantly reduces vascular wall disease in a murine model of type IIa hypercholesterolemia. Biomedicines 9: 1832.
- 71 Powell-Braxton L, Véniant M, Latvala RD, Hirano KI, Won WB, et al. (1998) A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med 4: 934–938.
- 72 Tanaka H, Iida Y, Iwaki T, Suzuki Y, Sano H, et al. (2018) Elevated plasma levels of LDL Cholesterol promote dissecting thoracic aortic aneurysms in angiotensin II-induced mice. Ann Vasc Surg 48: 204–213.
