Abstract
Optimizing the glucocorticoid dosage has been a major concern in classic 21OHD (21-hydroxylase deficiency) treatment, as it is essential to adjust it meticulously to the needs of the individual patient. Insufficient glucocorticoid treatment will cause adrenal insufficiency, including life-threatening adrenal crisis, while excess of androgen could cause precocious pubertal growth in children, virilization in female patients, and infertility in male and female adult patients. Meanwhile, overtreatment with glucocorticoids causes iatrogenic Cushing’s syndrome which could result in growth impairment, obesity, osteoporosis, and hypertension. The dilemma of 21OHD treatment is that glucocorticoid supplementation therapy at physiological dosage does not sufficiently suppress ACTH, consequently leading to adrenal androgen excess. Accordingly, the window for the appropriate glucocorticoid treatment would have to be substantially narrower than that of other types of adrenal insufficiency without androgen excess, such as adrenal hypoplasia. For the appropriate management of classic 21OHD, the physician has to be well versed in the physiology of the adrenal cortex, growth, and reproductive function. Comprehensive understanding of patients’ requirements according to their life stage and sex is essential. Furthermore, female patients with 46,XX need to be cared for as differences in sex development (DSD) with careful psychological management. In this review, we aimed to comprehensively summarize the current status of classic 21OHD treatment, including the initial treatment during the neonatal period, management of adrenal insufficiency, maintenance therapy of each life stage, and the importance of clinical management as DSD for 46,XX female patients. The recently developed agents, Chronocort, and Crinecerfont, are also discussed.
Introduction
Congenital adrenal hyperplasia (CAH) is caused by the loss or severe impairment of steroidogenic enzymes involved in cortisol biosynthesis. More than 90 percent of the cases result from 21-hydroxylase deficiency (21OHD) which is an inherited disorder caused by pathogenic variants in CYP21A2 [1, 2]. The prevalence of 21OHD is estimated to be 1:15,000–16,000 in the USA and Europe, and slightly lower in Japan at 1:18,000 without sex differences [1-4].
While the clinical spectrum of the disease ranges widely, it is mainly classified into two forms: classic and nonclassic. The classic form is further subdivided into two subtypes: the severest, salt wasting (SW) form and the simple virilizing (SV) form. The SW form is associated with cortisol and aldosterone deficiencies, in which neonates are likely to develop life-threatening adrenal crisis with severe hyponatremia and hyperkalemia. On the other hand, in the mildest, nonclassic type, the neonates are subclinical and develop hyperandrogenism during adolescence and adulthood in both sexes. Basically, nonclassic patients do not develop adrenal crisis [1-3, 5].
The introduction of newborn screening for congenital adrenal hyperplasia enabled to treat most patients with 21OHD with glucocorticoids from the neonatal period, remarkably improving the situation, including reducing inappropriate sex assignment in 46,XX patients, reducing mortality ratio during the neonatal—infant period, and increasing final height by suppressing bone age advancement [6-8]. However, glucocorticoid therapy and androgen excess have a “trade-off” relationship. Glucocorticoids have a strong potential for the adverse effect, iatrogenic Cushing’s syndrome, while insufficient glucocorticoid therapy leads to androgen excess [1, 2, 9, 10]. Optimizing the glucocorticoid therapy for 21OHD remains a challenging problem.
Recent studies have revealed that 21OHD patients are substantially at risk for metabolic syndrome in adulthood. Metabolic syndrome in 21OHD has been assumed to be due to long-term glucocorticoid therapy. Furthermore, the quality of life (QOL) of 21OHD patients in adulthood has been suggested to be substantially impaired, including sex issues in female patients [11-13]. Given the facts, the current standardized management of the glucocorticoid therapy for 21OHD children and adults remains suboptimal [9, 14].
We aimed to review the current management of 21OHD. In addition to the maintenance therapy of glucocorticoids, we also discussed the issues of initial treatment during the neonatal period, clinical management as DSD for 46,XX female 21OHD patients, testicular adrenal rest tumors (TART), and the new therapies including gene therapy.
General Issues of 21OHD Therapy
The treatment for classic 21OHD has two basic principles: 1. To supply glucocorticoids and mineralocorticoids, thus preventing adrenal insufficiency/crisis; 2. To reduce adrenal androgen excess, which seriously deteriorates the clinical outcomes of 21OHD patients through suppressing ACTH secretion from the pituitary gland (Fig. 1) [9, 10, 15]. With glucocorticoid therapy, the patients are able to lead an almost normal life [9, 10, 15].

Optimizing the glucocorticoid dosage has been a major concern of the 21OHD treatment, as it is essential to adjust it meticulously to the needs of the individual patient [2, 9, 10]. Insufficient glucocorticoid treatment will cause adrenal insufficiency, including life-threatening adrenal crisis, while excess of androgen would cause precocious pubertal growth in children, virilization in female patients, and infertility in male and female adult patients. On the other hand, overtreatment with glucocorticoids causes iatrogenic Cushing’s syndrome [2, 9, 10]. The dilemma of 21OHD treatment is that glucocorticoid supplementation therapy at physiological dosage does not sufficiently suppress ACTH, consequently leading to increased adrenal androgen levels [16, 17]. Since the physiological supplementation of glucocorticoid does not satisfactorily control the disease [10, 15, 18, 19], the window for appropriate glucocorticoid treatment is substantially narrower than that of other types of adrenal insufficiency without androgen excess and the approaches for maintenance therapy are not identical between the two conditions.
Another issue is that, in contrast to adrenal insufficiency, the problems with androgen excess differ by life stage and sex, and the glucocorticoid dosage has to be optimized according to their goal (Fig. 2) [10, 15]. During fetal period, external genitalia in 46,XX patients virilized by excess of androgen results in atypical genitalia that may not appear to be either male or female. In infancy–childhood–adolescence, androgen excess causes bone age advancement that leads to subsequent short stature in both sexes. In adulthood, suppressed gonadotropin by the negative feedback of excessive production of adrenal androgen results in infertility in both sexes [9]. In female patients, virilization due to insufficient control of androgen excess after birth causes numerous undesirable features, such as deepening of the voice, acne, hirsutism, increased muscle mass, and male pattern hair loss, seriously deteriorating the sexuality of the patients for a lifetime. Furthermore, virilized changes can be irreversible. Clinicians are thus required to understand the potential clinical problems of the patients according to their life stage and sex, as they seek to optimize their glucocorticoid treatment.

Glucocorticoid Therapy for Adrenal Insufficiency
Adrenal crisis, an acute type of adrenal insufficiency, is a life-threatening medical emergency, and one of the major goals of 21OHD treatment is to eradicate such lethal cases. Recent studies have revealed that adrenal crisis is still a menace. According to a Japanese nationwide study, 17.7% of 21OHD patients experienced at least one episode of hospital admission for adrenal crisis at the median age of 2 years. Another Italian national study for primary adrenal insufficiency (n = 803), in which 21OHD patients accounted for 85%, reported that the rate of adrenal crisis was 2.7 per 100 patient-years, and three patients died from adrenal crisis [20, 21]. Furthermore, even after a patient has recovered from adrenal crisis following glucocorticoid treatment, subsequent neurological comorbidities may occur [22-24]. Thus, the prevention of adrenal crisis is the primary goal of clinical management of 21OHD, and if adrenal crisis occurs, immediate administration of adequate glucocorticoid is required.
Because undiagnosed classic 21OHD patients are at the highest risk of adrenal crisis, newborn screening was introduced to Japan more than thirty years ago [4, 25-27]. There have been no reported neonate fatalities since the introduction of newborn screening, indicating that the primary goal of screening has been achieved. At the same time, a Tokyo screening study has shown that more than 30% of 21OHD neonates had already developed severe SW, which is defined by Na <130 mEq/L and K >7 mEq/L, upon arrival at medical hospitals [28]. Therefore, screening-positive neonates should be referred to hospitals as soon as possible. For the neonates that are already exhibiting clinical signs or symptoms, i.e., poor activity, dehydration, and shock, glucocorticoid should be administered immediately even before the definitive diagnosis of 21OHD. In such cases, collecting serum and urine samples for detailed endocrinological examination before glucocorticoid administration would be helpful for the later diagnosis of 21OHD.
Once the diagnosis of 21OHD is confirmed, patients and parents should be adequately instructed with regard to the management of adrenal crisis, following the sick day rule. In cases of substantial physical stress, such as febrile illness (>38.5°C), gastroenteritis, surgery under general anesthesia, and major trauma, extra glucocorticoid administration is required [10, 15]. When stress is severe, oral administration may not be sufficient for patients recovering from adrenal crisis, and intravenous or intramuscular injection of glucocorticoid might be required. In particular, patients in infancy and young childhood have a tendency to easily develop severe adrenal crisis [29]. Reducing opportunities for infection and securing access to emergency rooms are essential. Prescribing a glucocorticoid self-injection kit for emergency use would also help to greatly reduce a possibility of developing severe adrenal crisis. Wearing or carrying a medical identification card indicating adrenal insufficiency is also essential.
Maintenance Therapy of Glucocorticoid
Introduction
The primary goal of glucocorticoid maintenance therapy for 21OHD is to appropriately control androgen production from the adrenal gland without signs or symptoms of glucocorticoid excess or iatrogenic Cushing’s syndrome [2, 9, 15, 16, 19, 23].
Because the clinical problems caused by androgen excess differ according to life stage and gender, the definition of “appropriate” control would not be identical among 21OHD patients. Just automatically adjusting the dosage of glucocorticoid according to biomarkers, such as serum levels of 17OHP, would not lead to sufficient outcomes. The clinicians are required to understand the acceptable levels of androgen excess according to sex and the life-stage of the patients. As the amount of glucocorticoids for 21OHD treatment needed to lower androgen production is more than that which is physiologically required (estimated to be 5 to 7 mg/m2/day) [10, 18, 30], physicians should try to use the lowest possible dose of glucocorticoids for keeping androgen production at an acceptable level.
Types of glucocorticoid agent for maintenance therapy
Based on half-life, glucocorticoids are divided into three groups, which are represented by hydrocortisone, prednisolone, and dexamethasone. During infancy–childhood, the short-acting glucocorticoid, hydrocortisone, is recommended for maintenance therapy because of lowered risk of adverse effects caused by the more potent long-acting glucocorticoids, including growth impairment, obesity, and reduced bone mineral density. The longer-acting glucocorticoids, prednisolone and dexamethasone, are reported to have growth suppression effects of 15-fold and 70–80-fold, respectively, compared to hydrocortisone [10, 31, 32]. For adults, long-acting glucocorticoids have been preferred because of their more potent effects to suppress adrenal androgen by improving adherence. However, recent analysis has revealed that long-acting glucocorticoids, especially dexamethasone, adversely increase the risk of comorbidities, such as obesity and insulin resistance [14, 33]. Currently, hydrocortisone or prednisolone are preferred over dexamethasone [14, 33]. Furthermore, unlike hydrocortisone and prednisolone, dexamethasone evades degradation by the placenta and directly affects the fetus in pregnant women and avoiding dexamethasone use is recommended for pregnant women [10, 15].
Neonatal–early infancy
After therapy for acute adrenal crisis, glucocorticoid therapy should be shifted to maintenance therapy immediately (The initial treatment of an acute phase during neonatal period will be discussed in a latter section.). A recent report has suggested that the clinical outcomes between gradual shift and immediate shift to maintenance therapy would be identical, i.e., adrenal androgen was satisfactorily suppressed in the second years of life without any significant difference in anthropometric data [34]. Due to relative mineralocorticoid resistance and the anti-mineralocorticoid effects of elevated 17OHP in this period, neonates and young infants require the therapy for SW to all newborns with classic 21OHD [2, 35, 36]. It is especially important to note that a lower dose of hydrocortisone for maintenance therapy alone is not capable of improving SW.
Sufficient sodium supplementation plays a pivotal role in early infancy with classic 21OHD, and for efficient therapy for SW. In addition to fludrocortisone, oral NaCl supplementation is recommended [9, 34, 37]. Sodium expands the extracellular volume and the delivery of Na+ to the distal collecting tubules creates a favorable gradient for K+ excretion [38, 39]. The potency of the treatment is determined by the additive effect of fludrocortisone and NaCl intake, and the dosage of fludrocortisone may have to be changed according to the amount of NaCl intake [38, 39]. Indeed, in the extreme condition of aldosterone resistance, known as pseudohypoaldosteronism, a single treatment of high Na+ intake should efficiently improve hyponatremia and hyperkalemia [40].
For monitoring maintenance therapy during neonatal-early infancy, body weight gain is an excellent biomarker. The existence of glucocorticoid insufficiency or SW causes body weight loss, and appropriate body weight gain indicates that glucocorticoid insufficiency and SW are being properly managed [34]. Indeed, a recent report of newborn screening in Tokyo revealed that body weight loss is an excellent predictor for the diagnosis of 21OHD and the development of SW [28], suggesting that, without appropriate intervention, body weight will not increase properly in 21OHD patients during the neonatal period.
Childhood–puberty
The primary goal of the maintenance therapy during childhood–puberty is improving adult height by preventing bone age acceleration due to adrenal androgen and iatrogenic Cushing’s syndrome due to overtreatment (Figs. 3, 4). For monitoring the maintenance treatment, growth velocity, pubertal stage, and bone age advancement should be taken into account. Growth velocity is an especially good marker for both overtreatment and undertreatment (Figs. 3, 4). Androgen excess due to undertreatment increases growth velocity, while iatrogenic Cushing’s syndrome decreases growth velocity [41, 42]. Other physical findings, such as pubertal development and marked advancement of bone age, indicate androgen excess. Obesity with increased BMI SDs is suggestive of overtreatment. In female patients, clinical symptoms of androgen excess, such as hirsutism and acne, should be carefully monitored. However, when growth velocity, bone age, and pubertal stage are kept at normal, it is likely that androgen excess is properly under control.
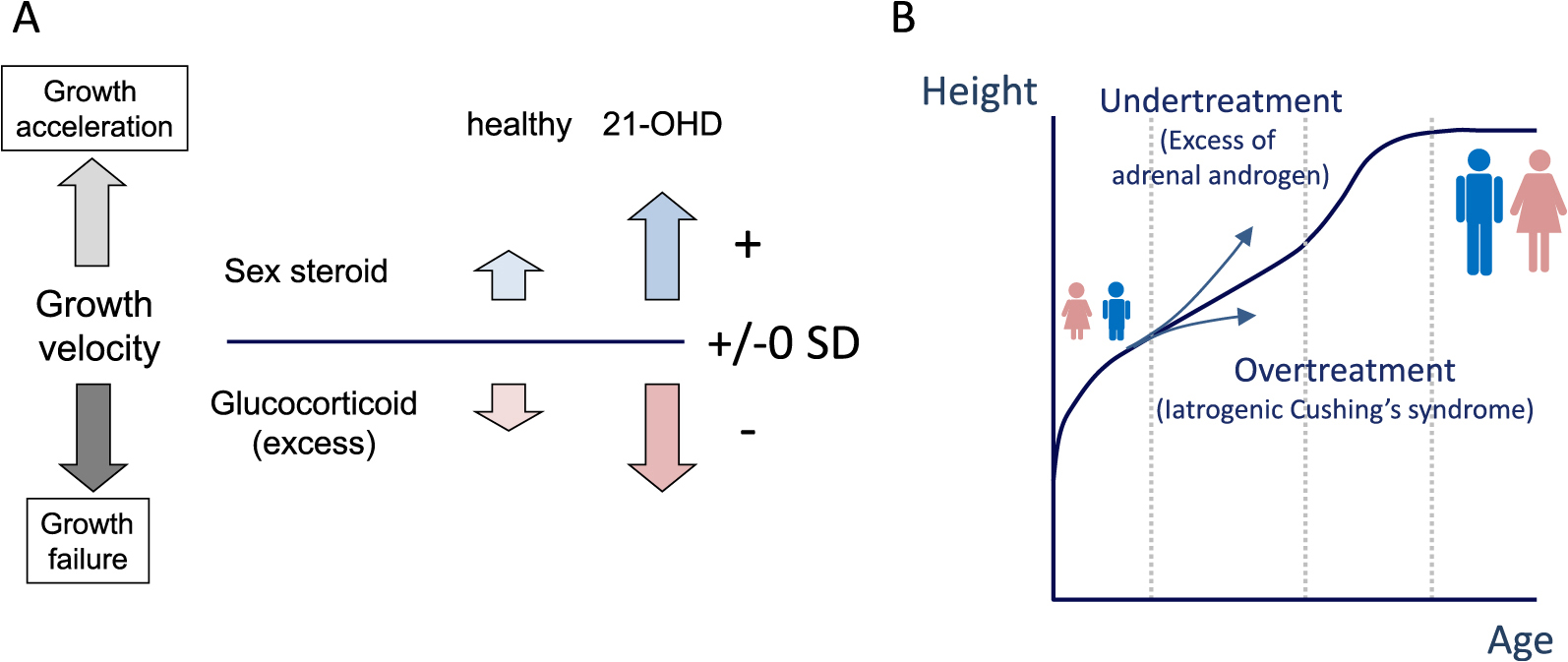
In addition to these physiological findings, using suitable biochemical markers is essential for monitoring glucocorticoid treatment. The most widely used biomarker is serum 17-OHP level [9, 10, 15, 43, 44]. The target range of serum 17-OHP levels just before taking the morning dose was suggested to be 4–12 ng/mL in both childhood and adulthood [2, 10, 45], although the range was not determined according to auxological data. Normal 17-OHP levels, on the other hand, would suggest overtreatment with glucocorticoids [2, 10].
A urine metabolite of 17-OHP, pregnanetriol, has been proposed for therapy monitoring, and, based on auxological data of 21OHD children, the optimal target range of urine pregnanetriol was suggested to be 1.2–2.1 mg/ m2/d and 2.2–3.3 mg/gCr in the first morning sample [41, 42]. Collecting the first morning urine before taking the morning medication would be more feasible than blood sampling [41, 42].
Serum androstenedione and testosterone levels can be used for monitoring therapy. The ratio of androstenedione/testosterone would especially differentiate the origin of androgen, i.e., whether it is adrenal cortex or gonads. In healthy females and males, the normal range is less than 2 and 0.2, respectively. Higher ratios (>4 in females and >0.5 in males) would indicate that the testosterone is mainly of adrenal origin [2].
We should be aware of the limitations of endocrinological data. The control indicated by endocrinological data is basically short-term and the value of hormones could vary at the conditions of sampling. We should interpret endocrinological data by repeating measurements accompanied by anthropometric data including bone age [9, 10, 15, 19].
An adequate sodium balance could decrease dosage of HCs by reducing levels of vasopressin which potentially increase ACTH release independently from corticotropin-releasing factor (CRF) [10], although its potential role in the physiology of CAH has yet to be established. Sensitivity to mineralocorticoid increases with age, leading to less requirement of fludrocortisone and sodium supplementation. Generally, sodium supplementation is not necessary beyond the infantile period. On the other hand, hypertension due to excessive fludrocortisone can sometimes be observed after infancy and even during early childhood, hence adequate monitoring of blood pressure and plasma renin activity is essential [37].
Adulthood
The primary goal of maintenance therapy during adulthood is to appropriately suppress adrenal androgen to maintain satisfactory QOL, and prevent comorbidities due to overtreatment, such as metabolic syndrome and iatrogenic Cushing’s syndrome [2, 9, 10, 15, 16, 19, 46] (Fig. 2).
During adulthood, two pathways of adrenal androgen excess affect the condition: the direct action of androgen, resulting in virilization, which is a serious concern in females; and an indirect effect through the negative feedback loop to suppress LH/FSH secretion, leading to hypogonadism in both sexes (Fig. 5B) [2, 9, 10, 15, 16, 19]. By suppressing ACTH secretion, glucocorticoid administration will prevent virilization and recover LH/FSH secretion (Fig. 5C) [2, 9, 10, 15, 16, 19].

Low levels LH/FSH suggest poor control of adrenal androgen excess and result in infertility in males and females (Figs. 5, 6) [2, 9]. In males, spermatogenesis depends on FSH and LH-induced testosterone, while adrenal androgen excess impairs spermatogenesis by inhibiting LH and FSH secretion. Seminiferous tubules, which are FSH-dependent, comprise the major volume of the testes, and the lower levels of FSH remarkably reduce their volume. In the absence of tumor mass, such as testicular adrenal rest tumor (TART), testicular volume (TV) is a valuable biomarker to predict fertility. In other words, 21OHD male patients with small TV are likely to be infertile due to insufficient control of adrenal androgen excess. From studies of infertile males, the potential cut-off value of TV to predict infertility would be 8–12 mL [47, 48]. In such cases, the basal level of LH and FSH would be less than that of the normal reference range in healthy adult males. To confirm the diagnosis of oligospermia/azoospermia, an examination of the semen is required.

In female patients, the menstrual cycle, including ovulation, is LH/FSH dependent, in other words LH/FSH insufficiency results in amenorrhea-oligomenorrhea. Regular menstruation in 21OHD female patients is thus suggestive of sufficient control of adrenal androgen excess [49]. Even though sufficient LH/FSH secretion is achieved, female 21OHD patients may develop one of the phenotypes overlapping with polycystic ovary syndrome (PCOS) with abnormal menstrual cycles [50, 51]. These clinical phenotypes can be explained by early exposure to androgens and progesterone oversecretion since the prenatal period [52, 53], as well as progesterone and androgen oversecretion during the follicular phase of the menstrual cycle [51, 52, 54].
Virilization can be a serious problem for female 21OHD patients. Physicians should carefully check for signs of hyperandrogenism, such as acne, hirsutism, balding, and deepening voice. Although race-specific normative ranges should be examined, the Ferriman-Gallwey scoring system is useful for systematic evaluation of hirsutism [55-59]. Grading systems for acne include quantity (mild, moderate, or severe), location, and quality (comedonal, inflammatory, or mixed) can be considered, but no grading scales have been universally agreed upon. If female 21OHD patients are depressed by hyperandrogenism despite continuously-maintained adherence, the dosage of glucocorticoid or a switch to a long-acting glucocorticoid would be an option, although careful monitoring for iatrogenic Cushing’s syndrome is required [46].
Just as adrenal androgen excess deteriorates the QOL in both sexes of 21OHD patients, so does iatrogenic Cushing’s syndrome. Deterioration in QOL has been reported to be associated with obesity and insulin resistance. In a meta-analysis, despite a greater potential to suppress adrenal androgen production, dexamethasone significantly increased the risk of higher BMI and lower bone mineral density than prednisolone and shorter-acting hydrocortisone [33, 60, 61]. Complete suppression of endogenous adrenal steroid secretion implies overtreatment, resulting in iatrogenic Cushing’s syndrome [60, 61]. Physicians should be aware of the trade-off between suppressing adrenal androgen excess and the risk of iatrogenic Cushing’s syndrome and should seek to optimize the glucocorticoid dosage for each patient according to their life stage and gender (Fig. 6) [46].
Taken together, the approach to maintenance therapy of 21OHD adults differs according to the life stage and gender of the patient. For example, a young woman who wants improved fertility might temporarily accept a high-dose regimen to increase her chances of conceiving (Fig. 6A line a) [46]. By contrast, a middle-aged man who has no requirement for fertility might want to receive physiological replacement of cortisol with hydrocortisone to reduce the risk of osteoporosis and cardiometabolic disorders (Fig. 6A line b) [46].
Other Topics of the 21OHD Treatment
Initial treatment during neonatal period
Japanese guidelines historically recommended the use of high-dose hydrocortisone (100–200 mg/m2/d) on initial treatment to suppress excessive adrenal androgen levels and to rapidly reduce hydrocortisone dosages when adrenal androgen levels reach target range [10, 62, 63]. A high initial high-dose hydrocortisone may help to normalize the elevated adrenal androgen levels rapidly, followed by sufficient control with a lower maintenance dose of glucocorticoid. In contrast, for securing the height at the age of 2–3 years, which is a strong predictor of adult height, European and American guidelines recommend lower dosage of glucocorticoid for initial treatment from 10–15 to 25 mg/m2/day [10, 15, 64]. In studies using initial treatment with low-dose hydrocortisone (9–15 mg/m2/d) and high-dose hydrocortisone (100–200 mg/m2/d), however, the linear growth of the first three years of life was comparable [34, 65], and bone age acceleration was not obvious under either regimen [34, 64, 65]. Further detailed analysis did not reveal any apparent advantage or disadvantage in using a higher dose of initial HC therapy for 21OHD in terms of suppression of adrenal androgen, body weight, body height, and BMI [34]. Accordingly, there is no clear evidence for the optimal dosage of glucocorticoids in initial treatment, and the current recommended initial dosage of HC in the neonatal period in Japan is 25–100 mg/m2/d [10]. The dosages and modes of administration should be adjusted individually based on the patient’s condition, such as severity of adrenal crisis, and clinical experience [10].
Clinical management as DSD for 46,XX female 21OHD patients
For the clinical management of female 21OHD patients, interdisciplinary teams for psychosocial problems specific to DSD should be involved during childhood–adolescence–adulthood [10, 15]. In addition to glucocorticoid and mineralocorticoid deficiency, 21OHD female patients have numerous clinical problems such as DSD. At birth, classic 46,XX female 21OHD patients have virilized external genitalia that do not have the typical appearance of female genitalia. Atypical genitalia of unknown cause would lead to difficulties of initial gender assignment, and the experience is often devastating for the patient’s parents [5, 10, 46]. Since most female-raised adolescents and adults with 46,XX 21OHD end up with a female gender identity [66, 67], it would be judicious to assign 46,XX 21OHD patients as female [10, 66, 67]. However, it inevitably means that female 21OHD patients will have to face two major clinical problems.
Exposure to androgen excess during fetal period results in atypical development of the external genitalia with variable extent of virilization in female 21OHD patients, requiring surgical restoration of functional anatomy. The principle of surgical therapy differs according to the patient’s particular social background and country of origin [68]. In Japan, plastic surgery is generally performed during early childhood, before the age of two years, when gender development remains immature [10]. On the other hand, in western countries, the timing of surgical therapy has been debated [68] since patient participation has to be considered for the decision-making process since the consensus statement of DSD [69]. QOL and psychosexual behavior in female 21OHD patients are seriously affected by genital surgery and its consequences [67, 70-72]. The postoperative complications of genital surgery, such as vaginal stenosis, labial scarring, urethra-vaginal fistulae, urinary incontinence, and recurrent urinary tract infection, require further optimization of continuous care for the patients [73]. Needless to say, plastic surgery should only be performed in centers with a staff of experienced pediatric surgeons/urologists and pediatric endocrinologists [10, 15].
Additionally, as female 21OHD patients are exposed to the threat of androgen excess for the rest of their lives, these clinical problems take a serious toll on their psychological health [74]. Girls and women with CAH have an increased risk of reaction to extreme stress, adjustment disorders, and alcohol abuse compared with those without the disorder, and the risk is highest among the patients with the severest type [74, 75].
Androgen suppression is obviously important for achieving satisfactory QOL in 21OHD female adults. Desirable androgen suppression in 21OHD female adults has to be precisely treated according to life stage. This issue remains an open question for the future study of 21OHD.
Testicular adrenal rest tissue (TART) and ovarian adrenal rest tissue (OART)
Adrenal rest tissue is ectopic adrenal tissue and is most often associated with the testes because steroidogenic cells in the adrenal glands and gonads develop from the same gonadal primordium, the urogenital ridge. Testicular adrenal rest tissue (TART) is one of the potential factors to cause male subfertility [76-78]. TART blockage of the seminiferous tubules would result in azoospermia and failure of androgen synthesis by damaging the Leydig cells. Undertreated patients have the tendency to develop TART, potentially due to increased ACTH which would drive TART growth, although a clear correlation between TART development and plasma levels of ACTH has yet to be established [79, 80]. TART is prevalent in male patients with classic 21OHD, and regular monitoring by ultrasonography in males after 10 years of age is required [9, 10, 15]. Optimization of glucocorticoid therapy would reduce the size of the TART in the early stage, preventing progressive enlargement which would deteriorate testicular function [10, 15].
On the other hand, a few female 21OHD cases have been reported with ovarian mass due to ectopic adrenal tissue, i.e., ovarian adrenal rest tissue (OART) [81]. Despite its extremely low prevalence, poor adherence is a common feature of 21OHD female patients with OART, and close follow-up for OART is necessary for those patients [81].
Future Direction
New therapies
The majority of the poor health outcomes in 21OHD patients result from the difficulty of maintaining a balance between preventing iatrogenic Cushing’s syndrome and the sufficient attenuation of adrenal androgen excess (Fig. 6) [9, 10, 15, 46]. The pharmacokinetics of the currently available glucocorticoid agents is totally different from the physiological circadian rhythm of serum cortisol levels (Fig. 7A) [9, 44]. Thus, it is extremely challenging to replicate the physiological circadian rhythm by using the currently available glucocorticoid agents.

To date, numerous therapeutic approaches have been examined for solving the problem, and one of the candidates is a newly modified-release formulation of hydrocortisone called Efmody® (development name Chronocort, Diurnal Europe B.V, The Netherlands) [9]. Chronocort is a multi-particulate formulation in which hydrocortisone is embedded, enabling delayed release of hydrocortisone and sustained absorption (Fig. 7B) [82-85]. By administration at bedtime and early morning, Chronocort fairly replicates the physiological circadian rhythm of cortisol [82, 83]. In phase 2 and 3 clinical trials, Chronocort, at a lower dose than the standard treatment, improved control of 21OHD patients with reduced AUC and amplitude of 17OHP [85, 86]. The improved disease control was observed despite a reduction in hydrocortisone dose by 33%, to doses typically used for adrenal replacement therapy (15–25 mg/day) [86]. The data imply that the dilemma of 21OHD therapy might one day be solved by using Chronocort.
Another target of the new therapy is CRF. Crinecerfont (NBI-74788) is a recently developed non-peptide CRF1 receptor antagonist which suppresses androgen synthesis from the adrenal gland in 21OHD patients by inhibiting ACTH production [87]. Theoretically, Crinecerfont allows for glucocorticoids to be administered at more physiologic doses, reducing the risk of iatrogenic Cushing’s syndrome [87]. In an open-label phase 2 clinical trial, Crinecerfont was revealed to reduce ACTH and 17OHP levels in a dose-dependent manner [87]. Currently, efficacy, safety, and tolerability are being evaluated in a phase 3 study [88].
Gene therapy for a monogenic disorder could improve QOL of the patients without the burden of daily disease management and its complications, and 21OHD is no exception. The recent rapid development of AAV (adeno-associatedvirus)-mediated gene targeting system has enabled us to attempt a human gene therapy of 21OHD. AAV systems have successfully introduced the Cyp21/CYP21A2 gene into 21OHD model mice and iPS cells from 21OHD patients, respectively [89-91]. Despite the limitation of the outcomes, the data suggest that disruption of the Cyp21/CYP21A2 gene can be treated with extra-adrenal gene induction.
Summary
In this review, we outlined the current aspect of the 21OHD treatment, including initial treatment during the neonatal period, management of adrenal insufficiency, maintenance therapy at each life stage, the importance of clinical management of 46,XX female 21OHD patients as DSD, and the new glucocorticoid drug, Chronocort. Although the treatment of patients with 21OHD has improved over time, optimizing the treatment is still extremely challenging. In addition to deep knowledge of the endocrinological physiology of the adrenal cortex, growth, and reproductive function, comprehensive understanding of patients’ requirement according to their life stage and gender is essential for appropriate management of 21OHD. Female patients with 46,XX should also be cared for as DSD, and careful psychological management is essential. Recent advances in developing therapies, including Chronocort and Crinecerfont, are expected to significantly improve the patient outcomes. Further research is required to monitor the health outcomes in patients with 21OHD.
Acknowledgements
The present study was partly supported by a grant from the Ministry of Health, Labour and Welfare of Japan (20FC1020), and Japan Agency for Medical Research and Development (AMED) (#23ek0109630h0001).
We would like to express our sincere thanks to Dr. Tomonobu Hasegawa (Keio University) for valuable suggestions and comments.
Disclosure
None of the authors have any potential conflicts of interest associated with this research.
References
- 1 Speiser PW, White PC (2003) Congenital adrenal hyperplasia. N Engl J Med 349: 776–788.
- 2 Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, et al. (2022) Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev 43: 91–159.
- 3 White PC, New MI, Dupont B (1987) Congenital adrenal hyperplasia. (1). N Engl J Med 316: 1519–1524.
- 4 Tsuji A, Konishi K, Hasegawa S, Anazawa A, Onishi T, et al. (2015) Newborn screening for congenital adrenal hyperplasia in Tokyo, Japan from 1989 to 2013: a retrospective population-based study. BMC Pediatr 15: 209.
- 5 White PC, Speiser PW (2000) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 21: 245–291.
- 6 Therrell BL Jr, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, et al. (1998) Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics 101: 583–590.
- 7 Heather NL, Seneviratne SN, Webster D, Derraik JG, Jefferies C, et al. (2015) Newborn screening for congenital adrenal hyperplasia in New Zealand, 1994–2013. J Clin Endocrinol Metab 100: 1002–1008.
- 8 Balsamo A, Cicognani A, Baldazzi L, Barbaro M, Baronio F, et al. (2003) CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. J Clin Endocrinol Metab 88: 5680–5688.
- 9 Mallappa A, Merke DP (2022) Management challenges and therapeutic advances in congenital adrenal hyperplasia. Nat Rev Endocrinol 18: 337–352.
- 10 Ishii T, Kashimada K, Amano N, Takasawa K, Nakamura-Utsunomiya A, et al. (2022) Clinical guidelines for the diagnosis and treatment of 21-hydroxylase deficiency (2021 revision). Clin Pediatr Endocrinol 31: 116–143.
- 11 Charmandari E, Chrousos GP (2006) Metabolic syndrome manifestations in classic congenital adrenal hyperplasia: do they predispose to atherosclerotic cardiovascular disease and secondary polycystic ovary syndrome? Ann N Y Acad Sci 1083: 37–53.
- 12 Bouvattier C, Esterle L, Renoult-Pierre P, de la Perriere AB, Illouz F, et al. (2015) Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. A french national survey. J Clin Endocrinol Metab 100: 2303–2313.
- 13 Rosenbaum D, Gallo A, Lethielleux G, Bruckert E, Levy BI, et al. (2019) Early central blood pressure elevation in adult patients with 21-hydroxylase deficiency. J Hypertens 37: 175–181.
- 14 Han TS, Krone N, Willis DS, Conway GS, Hahner S, et al. (2013) Quality of life in adults with congenital adrenal hyperplasia relates to glucocorticoid treatment, adiposity and insulin resistance: United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE). Eur J Endocrinol 168: 887–893.
- 15 Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, et al. (2018) Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 103: 4043–4088.
- 16 Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, et al. (2010) Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab 95: 5110–5121.
- 17 Finkielstain GP, Kim MS, Sinaii N, Nishitani M, Van Ryzin C, et al. (2012) Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 97: 4429–4438.
- 18 Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH (2004) Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab 89: 281–287.
- 19 Joint LWPES/ ESPE CAH Working Group (2002) Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab 87: 4048–4053.
- 20 Ishii T, Adachi M, Takasawa K, Okada S, Kamasaki H, et al. (2018) Incidence and characteristics of adrenal crisis in children younger than 7 years with 21-hydroxylase deficiency: a nationwide survey in Japan. Horm Res Paediatr 89: 166–171.
- 21 Capalbo D, Moracas C, Cappa M, Balsamo A, Maghnie M, et al. (2021) Primary adrenal insufficiency in childhood: data from a large nationwide cohort. J Clin Endocrinol Metab 106: 762–773.
- 22 Nass R, Heier L, Moshang T, Oberfield S, George A (1997) Magnetic resonance imaging in the congenital adrenal hyperplasia population: increased frequency of white-matter abnormalities and temporal lobe atrophy. J Child Neurol 12: 181–186.
- 23 Ishii T, Tajima T, Kashimada K, Mukai T, Tanahashi Y, et al. (2020) Clinical features of 57 patients with lipoid congenital adrenal hyperplasia: criteria for nonclassic form revisited. J Clin Endocrinol Metab 105: dgaa557.
- 24 Abe Y, Sakai T, Okumura A, Akaboshi S, Fukuda M, et al. (2016) Manifestations and characteristics of congenital adrenal hyperplasia-associated encephalopathy. Brain Dev 38: 638–647.
- 25 Tsuji-Hosokawa A, Kashimada K (2021) Thirty-year lessons from the newborn screening for congenital adrenal hyperplasia (CAH) in Japan. Int J Neonatal Screen 7: 36.
- 26 Suwa S (1994) Nationwide survey of neonatal mass-screening for congenital adrenal hyperplasia in Japan. Screening 3: 141–151.
- 27 Suwa S (1994) Congenital adrenal hyperplasia. Shouni-Naika 26: 1967–1972 (In Japanese).
- 28 Gau M, Konishi K, Takasawa K, Nakagawa R, Tsuji-Hosokawa A, et al. (2020) The progression of salt wasting and the body weight change during the first two weeks of life in classical 21-hydroxylase deficiency patients. Clin Endocrinol (Oxf) 94: 229–236.
- 29 Reisch N, Willige M, Kohn D, Schwarz HP, Allolio B, et al. (2012) Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur J Endocrinol 167: 35–42.
- 30 Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD (1993) Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 76: 1505–1510.
- 31 Punthakee Z, Legault L, Polychronakos C (2003) Prednisolone in the treatment of adrenal insufficiency: a re-evaluation of relative potency. J Pediatr 143: 402–405.
- 32 Rivkees SA, Crawford JD (2000) Dexamethasone treatment of virilizing congenital adrenal hyperplasia: the ability to achieve normal growth. Pediatrics 106: 767–773.
- 33 Whittle E, Falhammar H (2019) Glucocorticoid regimens in the treatment of congenital adrenal hyperplasia: a systematic review and meta-analysis. J Endocr Soc 3: 1227–1245.
- 34 Saito Y, Takasawa K, Ga M, Yamauchi T, Nakagawa R, et al. (2021) Adrenal suppression and anthropometric data at two years of age was not influenced by the initial hydrocortisone dose in patients with 21-hydroxylase deficiency. Clin Pediatr Endocrinol 30: 155–161.
- 35 Mooij CF, Parajes S, Pijnenburg-Kleizen KJ, Arlt W, Krone N, et al. (2015) Influence of 17-hydroxyprogesterone, progesterone and sex steroids on mineralocorticoid receptor transactivation in congenital adrenal hyperplasia. Horm Res Paediatr 83: 414–421.
- 36 Quinkler M, Meyer B, Bumke-Vogt C, Grossmann C, Gruber U, et al. (2002) Agonistic and antagonistic properties of progesterone metabolites at the human mineralocorticoid receptor. Eur J Endocrinol 146: 789–799.
- 37 Neumann U, van der Linde A, Krone RE, Krone NP, Guven A, et al. (2022) Treatment of congenital adrenal hyperplasia in children aged 0–3 years: a retrospective multicenter analysis of salt supplementation, glucocorticoid and mineralocorticoid medication, growth and blood pressure. Eur J Endocrinol 186: 587–596.
- 38 Wright FS (1977) Sites and mechanisms of potassium transport along the renal tubule. Kidney Int 11: 415–432.
- 39 Good DW, Wright FS (1979) Luminal influences on potassium secretion: sodium concentration and fluid flow rate. Am J Physiol 236: F192–F205.
- 40 Belot A, Ranchin B, Fichtner C, Pujo L, Rossier BC, et al. (2008) Pseudohypoaldosteronisms, report on a 10-patient series. Nephrol Dial Transplant 23: 1636–1641.
- 41 Izawa M, Aso K, Higuchi A, Ariyasu D, Hasegawa Y (2007) Pregnanetriol in the range of 1.2–2.1 mg/m2/day as an index of optimal control in CYP21A2 deficiency. Clin Pediatr Endocrinol 16: 45–52.
- 42 Izawa M, Aso K, Higuchi A, Ariyasu D, Hasegawa Y (2008) The Range of 2.2–3.3 mg/gCr of Pregnanetriol in the first morning urine sample as an index of optimal control in CYP21 deficiency. Clin Pediatr Endocrinol 17: 75–80.
- 43 New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, et al. (1983) Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab 57, 320–326.
- 44 Charmandari E, Matthews DR, Johnston A, Brook CG, Hindmarsh PC (2001) Serum cortisol and 17-hydroxyprogesterone interrelation in classic 21-hydroxylase deficiency: is current replacement therapy satisfactory? J Clin Endocrinol Metab 86: 4679–4685.
- 45 Pham-Huu-Trung MT, Gourmelen M, Girard F (1973) The simultaneous assay of cortisol and 17alpha-hydroxyprogesterone in the plasma of patients with congenital adrenal hyperplasia. Acta Endocrinol (Copenh) 74: 316–330.
- 46 Han TS, Walker BR, Arlt W, Ross RJ (2014) Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol 10: 115–124.
- 47 Kliesch S, Behre HM, Nieschlag E (1995) Recombinant human follicle-stimulating hormone and human chorionic gonadotropin for induction of spermatogenesis in a hypogonadotropic male. Fertil Steril 63, 1326–1328.
- 48 Lin J, Mao J, Wang X, Ma W, Hao M, et al. (2019) Optimal treatment for spermatogenesis in male patients with hypogonadotropic hypogonadism. Medicine (Baltimore) 98: e16616.
- 49 Bulun S (2020) Physiology and pathology of the female reproductive axis. In: Melmed S, Koenig RJ, Rosen CJ, Auchus RJ, Goldfine AB (eds) Williams textbook of endocrinology (14th). Elsevier, Philadelphia, USA: 574–641.
- 50 Helleday J, Siwers B, Ritzen EM, Carlstrom K (1993) Subnormal androgen and elevated progesterone levels in women treated for congenital virilizing 21-hydroxylase deficiency. J Clin Endocrinol Metab 76: 933–936.
- 51 Holmes-Walker DJ, Conway GS, Honour JW, Rumsby G, Jacobs HS (1995) Menstrual disturbance and hypersecretion of progesterone in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf) 43: 291–296.
- 52 Barnes RB, Rosenfield RL, Ehrmann DA, Cara JF, Cuttler L, et al. (1994) Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab 79: 1328–1333.
- 53 Belgorosky A, Chahin S, Rivarola MA (1996) Elevation of serum luteinizing hormone levels during hydrocortisone treatment in infant girls with 21-hydroxylase deficiency. Acta Paediatr 85: 1172–1175.
- 54 Bachelot A, Chakhtoura Z, Plu-Bureau G, Coudert M, Coussieu C, et al. (2012) Influence of hormonal control on LH pulsatility and secretion in women with classical congenital adrenal hyperplasia. Eur J Endocrinol 167: 499–505.
- 55 Ferriman D, Gallwey JD (1961) Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 21: 1440–1447.
- 56 Ferriman D, Purdie AW (1965) Association of Oligomenorrhoea, Hirsuties, and Infertility. Br Med J 2: 69–72.
- 57 Schernthaner-Reiter MH, Baumgartner-Parzer S, Egarter HC, Krebs M, Kautzky-Willer A, et al. (2019) Influence of genotype and hyperandrogenism on sexual function in women with congenital adrenal hyperplasia. J Sex Med 16: 1529–1540.
- 58 Escobar-Morreale HF, Carmina E, Dewailly D, Gambineri A, Kelestimur F, et al. (2012) Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 18: 146–170.
- 59 Ilagan MKCC, Paz-Pacheco E, Totesora DZ, Clemente-Chua LR, Jalique JRK (2019) The modified Ferriman-Gallwey score and hirsutism among Filipino Women. Endocrinol Metab (Seoul) 34: 374–381.
- 60 Han TS, Stimson RH, Rees DA, Krone N, Willis DS, et al. (2013) Glucocorticoid treatment regimen and health outcomes in adults with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 78: 197–203.
- 61 Seraphim CE, Frassei JS, Pessoa BS, Scalco RC, Miranda MC, et al. (2019) Impact of long-term dexamethasone therapy on the metabolic profile of patients with 21-hydroxylase deficiency. J Endocr Soc 3: 1574–1582.
- 62 Saisho S, Yokota I, Kusuda S, Tachibana K, Igarashi Y (1999) Guidelines for diagnosis of 21-hydropxylase deficiency. J Jpn Pediatr Soc 103: 695–697 (In Japanese).
- 63 Takasawa K, Ono M, Miyai, K, Matsubara Y, Takizawa F, et al. (2012) Initial high dose hydrocortisone (HDC) treatment for 21-hydroxylase deficiency (21-OHD) does not affect linear growth during the first three years of life. Endocr J 59: 1001–1006.
- 64 Bonfig W, Schmidt H, Schwarz HP (2011) Growth patterns in the first three years of life in children with classical congenital adrenal hyperplasia diagnosed by newborn screening and treated with low doses of hydrocortisone. Horm Res Paediatr 75: 32–37.
- 65 Takasawa K, Ono M, Hijikata A, Matsubara Y, Katsumata N, et al. (2014) Two novel HSD3B2 missense mutations with diverse residual enzymatic activities for Delta5-steroids. Clin Endocrinol (Oxf) 80: 782–789.
- 66 Dessens AB, Slijper FM, Drop SL (2005) Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav 34: 389–397.
- 67 Meyer-Bahlburg HF, Dolezal C, Baker SW, Ehrhardt AA, New MI (2006) Gender development in women with congenital adrenal hyperplasia as a function of disorder severity. Arch Sex Behav 35: 667–684.
- 68 Hebenstreit D, Ahmed SF, Krone N, Krall C, Bryce J, et al. (2021) Surgical practice in girls with congenital adrenal hyperplasia: an international registry study. Sex Dev 15: 229–235.
- 69 Hughes IA, Houk C, Ahmed SF, Lee PA, Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group (2006) Consensus statement on management of intersex disorders. J Pediatr Urol 2: 148–162.
- 70 Nordenstrom A, Frisen L, Falhammar H, Filipsson H, Holmdahl G, et al. (2010) Sexual function and surgical outcome in women with congenital adrenal hyperplasia due to CYP21A2 deficiency: clinical perspective and the patients’ perception. J Clin Endocrinol Metab 95: 3633–3640.
- 71 Nordenskjold A, Holmdahl G, Frisen L, Falhammar H, Filipsson H, et al. (2008) Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. J Clin Endocrinol Metab 93: 380–386.
- 72 Meyer-Bahlburg HFL, Khuri J, Reyes-Portillo J, Ehrhardt AA, New MI (2018) Stigma associated with classical congenital adrenal hyperplasia in women’s sexual lives. Arch Sex Behav 47: 943–951.
- 73 Baskin A, Wisniewski AB, Aston CE, Austin P, Chan YM, et al. (2020) Post-operative complications following feminizing genitoplasty in moderate to severe genital atypia: results from a multicenter, observational prospective cohort study. J Pediatr Urol 16: 568–575.
- 74 Oner O, Aycan Z, Tiryaki T, Soy D, Cetinkaya E, et al. (2009) Variables related to behavioral and emotional problems and gender typed behaviors in female patients with congenital adrenal hyperplasia. J Pediatr Endocrinol Metab 22: 143–151.
- 75 Engberg H, Butwicka A, Nordenstrom A, Hirschberg AL, Falhammar H, et al. (2015) Congenital adrenal hyperplasia and risk for psychiatric disorders in girls and women born between 1915 and 2010: a total population study. Psychoneuroendocrinology 60: 195–205.
- 76 Claahsen-van der Grinten HL, Otten BJ, Hermus AR, Sweep FC, Hulsbergen-van de Kaa CA (2008) Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertil Steril 89: 597–601.
- 77 Cabrera MS, Vogiatzi MG, New MI (2001) Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 86: 3070–3078.
- 78 Stikkelbroeck NM, Otten BJ, Pasic A, Jager GJ, Sweep CG, et al. (2001) High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab 86: 5721–5728.
- 79 Martinez-Aguayo A, Rocha A, Rojas N, Garcia C, Parra R, et al. (2007) Testicular adrenal rest tumors and Leydig and Sertoli cell function in boys with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 92: 4583–4589.
- 80 Reisch N, Rottenkolber M, Greifenstein A, Krone N, Schmidt H, et al. (2013) Testicular adrenal rest tumors develop independently of long-term disease control: a longitudinal analysis of 50 adult men with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency. J Clin Endocrinol Metab 98: E1820–E1826.
- 81 Koren R, Koren S, Khashper A, Benbassat C, Pekar-Zlotin M, et al. (2021) Ovarian adrenal rest tumor in congenital adrenal hyperplasia: Is medical treatment the first line option? Arch Endocrinol Metab 65: 841–845.
- 82 Verma S, Vanryzin C, Sinaii N, Kim MS, Nieman LK, et al. (2010) A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 72: 441–447.
- 83 Whitaker M, Debono M, Huatan H, Merke D, Arlt W, et al. (2014) An oral multiparticulate, modified-release, hydrocortisone replacement therapy that provides physiological cortisol exposure. Clin Endocrinol (Oxf) 80: 554–561.
- 84 Newell-Price J, Whiteman M, Rostami-Hodjegan A, Darzy K, Shalet S, et al. (2008) Modified-release hydrocortisone for circadian therapy: a proof-of-principle study in dexamethasone-suppressed normal volunteers. Clin Endocrinol (Oxf) 68: 130–135.
- 85 Mallappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, et al. (2015) A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 100: 1137–1145.
- 86 Prete A, Auchus RJ, Ross RJ (2021) Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur J Endocrinol 186: R1–R14.
- 87 Auchus RJ, Sarafoglou K, Fechner PY, Vogiatzi MG, Imel EA, et al. (2022) Crinecerfont lowers elevated hormone markers in adults with 21-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab 107: 801–812.
- 88 National Institutes of Health (2023) Global Safety and Efficacy Registration Study of Crinecerfont for Congenital Adrenal Hyperplasia (CAHtalyst). Vol. 2023. https://clinicaltrials.gov/ct2/show/NCT04806451 accessed on January 15, 2023.
- 89 Perdomini M, Dos Santos C, Goumeaux C, Blouin V, Bougneres P (2017) An AAVrh10-CAG-CYP21-HA vector allows persistent correction of 21-hydroxylase deficiency in a Cyp21(–/–) mouse model. Gene Ther 24: 275–281.
- 90 Naiki Y, Miyado M, Horikawa R, Katsumata N, Onodera M, et al. (2016) Extra-adrenal induction of Cyp21a1 ameliorates systemic steroid metabolism in a mouse model of congenital adrenal hyperplasia. Endocr J 63: 897–904.
- 91 Naiki Y, Miyado M, Shindo M, Horikawa R, Hasegawa Y, et al. (2022) Adeno-associated virus-mediated gene therapy for patients’ fibroblasts, induced pluripotent stem cells, and a mouse model of congenital adrenal hyperplasia. Hum Gene Ther 33: 801–809.

