REVIEW
Cardiovascular complications in insulin resistance and endocrine diseases
2023 Volume 70 Issue 3 Pages 249-257

Details
2023 Volume 70 Issue 3 Pages 249-257
Cerebrovascular diseases, such as stroke and cardiovascular disease, are one of the leading causes of death in Japan. Type 2 diabetes is the most common form of diabetes and an important risk factor for these diseases. Among various pathological conditions associated with type 2 diabetes, insulin resistance has already been reported to be an important risk factor for diabetic complications. The major sites of insulin action in glucose metabolism in the body include the liver, skeletal muscle, and adipose tissue. However, insulin signaling molecules are also constitutively expressed in vascular endothelial cells, vascular smooth muscle, and monocytes/macrophages. Forkhead box class O family member proteins (FoxOs) of transcription factors play important roles in regulating glucose and lipid metabolism, oxidative stress response and redox signaling, and cell cycle progression and apoptosis. FoxOs in vascular endothelial cells strongly promote arteriosclerosis by suppressing nitric oxide production, enhancing inflammatory response, and promoting cellular senescence. In addition, primary aldosteronism and Cushing’s syndrome are known to have adverse effects on the cardiovascular system, apart from hypertension, diabetes, and dyslipidemia. In the treatment of endocrine disorders, hormonal normalization by surgical treatment and receptor antagonists play an important role in preventing cardiovascular complications.
Cerebrovascular disease, including stroke and cardiovascular disease (CVD), are the leading causes of death in Japan, and accounted for 23.2% of the total deaths in 2018. The major causes of the need for long-term care in Japan are also cerebrovascular disease (16.1%) and CVD (4.5%), which account for more than 20% of the total deaths. Medical expenses for cerebrovascular and CVD account for approximately 20% of the total, which is the highest by injury/illness classification.
Type 2 diabetes is the most common type of diabetes and an important risk factor for atherosclerotic diseases such as stroke, ischemic heart disease, and peripheral arterial disease. The risk of these diseases is predicted to be 2–4 times higher in patients with type 2 diabetes than those without [1-4]. According to the “National Health and Nutrition Survey” released in 2019 by the Ministry of Health, Labour and Welfare in Japan, 10.8% of women and 19.7% of men are estimated as strongly suspected of having diabetes. In addition to the lower insulin secretion capacity of Japanese people than that of western people as a genetic factor, insulin resistance (a condition in which insulin is less effective) has developed exponentially owing to obesity and visceral fat accumulation caused by environmental factors such as the rapid westernization of diet and a lack of exercise [5, 6].
Moreover, some endocrine disorders, including primary aldosteronism (PA) and Cushing’s syndrome (CS), are known to exert deleterious cardiovascular effects independent of hypertension, diabetes, and dyslipidemia. In addition, excessive hormones themselves directly affect the cardiovascular system. Therefore, hormonal normalization by surgical treatment and receptor antagonists play an important role in preventing cardiovascular complications.
This review focuses on molecular mechanisms by which insulin resistance leads to cardiovascular complications, which were suggested by findings from clinical research and genetically engineered mice. It also deals with cardiovascular complications associated with endocrine disorders, with reference to basic findings and evidence from clinical studies.
Among the various conditions associated with type 2 diabetes, insulin resistance is a major risk factor for diabetic complications. According to epidemiological studies, such as UKPDS 33 [7] and 35 [8], the Kumamoto study [9, 10], and the Funagata study [4], among others, microvascular complications associated with type 2 diabetes are closely related to the development of long-term persistent hyperglycemia, while macrovascular complications such as atherosclerosis are caused by impaired glucose tolerance in the prediabetic stage.
There is a strong correlation between the risk of developing CVD and insulin resistance [11]. Investigators in the Insulin Resistance Atherosclerosis Study (IRAS) showed a direct correlation between insulin resistance and atherosclerosis [12] and a follow-up prospective study in a cohort of 2,938 patients reported insulin resistance as an important risk factor for CVD [13]. A meta-analysis of 65 studies including 516,325 participants, revealed that insulin resistance, evaluated by HOMA index, was a strong predictor for CVD [11]. Using the Archimedes model and a population representative of young nondiabetic adults aged 20–30 years, the authors concluded that preventing insulin resistance could avoid approximately 42% of the myocardial infarctions in the participants during a simulated follow-up period of 60 years [14]. A wealth of studies also supported the notion that CVD was related to insulin resistance [15-18]. Several molecular mechanisms contribute to the association between insulin resistance and CVD [19-22]. These mechanisms include insulin resistance, vascular function, hypertension, and macrophage accumulation in the development of atherosclerosis [19].
Although the primary organs of action of insulin in systemic glucose metabolism are the liver, skeletal muscle, and adipose tissue, insulin receptors (IRs) and their downstream insulin signaling-related molecules are constitutively expressed in vascular endothelial cells [23-25], vascular smooth muscle [26], and monocytes/macrophages [27]. The physiological role of the IR and its downstream insulin signaling-related molecules have been the focus of much attention since the 1990s. It has become clear that insulin promotes phosphorylation of endothelial nitric oxide synthase (eNOS) in an Akt- and PI3 kinase-dependent manner in vascular endothelial cells [24, 28], further establishing the relationship between insulin signaling and vascular function in vascular endothelial cells. It has also been reported that obese (ob/ob) rats develop insulin resistance in blood vessels, liver, skeletal muscle, and adipose tissue [23]. The study of the mechanisms of insulin signaling in blood vessels is beginning to receive increasing attention in order to elucidate its correlation with pathological conditions such as atherosclerosis.
Insulin signaling in vascular endothelial cellsVascular endothelial cells are a single layer of epithelial cells lining the vessel wall. In addition to acting as a barrier between the vascular lumen and the arterial wall, vascular endothelial cells release a variety of vasoactive factors and regulate many functions, including vasoconstriction, cell adhesion, platelet adhesion and aggregation, inflammation, and cell proliferation. Atherosclerosis is accompanied by morphological changes in the blood vessels (occlusion, stenosis, enlargement, rupture, and dissection). Abnormal vascular endothelial cell function preceding these changes is considered an early lesion of atherosclerosis. Crossbreeding vascular endothelial cell-specific IR-deficient mice with apolipoprotein E (ApoE)-deficient mice promotes atherosclerosis. However, the glucose tolerance, insulin levels, lipids, and blood pressure do not differ from those in controls [25]. In vascular endothelial cell of control ApoE-deficient mice, insulin phosphorylates Ser1177 of eNOS (eNOS activation) and suppresses expression of vascular cell adhesion molecule (VCAM)-1, which has been shown to exert anti-atherosclerotic effects.
In vascular endothelial cell-specific IR-deficient mice, these effects are reduced, while nitric oxide (NO)-induced vasodilation is suppressed and VCAM-1-dependent leukocyte adhesion is enhanced. Furthermore, it has been reported that vascular endothelial cell-specific IR dominant-negative transgenic mice have attenuated vasorelaxation responses independent of systemic glucose metabolism and blood pressure [29]. Phosphorylation of eNOS is decreased in Akt1-deficient mice, a member of the Akt subfamily of downstream molecules of insulin signaling, and accelerated atherosclerosis is observed in ApoE-deficient mice [30]. On the other hand, mice overexpressing insulin receptor substrate 1 (IRS1) specifically in vascular endothelial cells show inhibition of atherosclerosis [31]. These reports suggest that insulin signaling in vascular endothelial cells prevents atherosclerosis, while insulin resistance almost consistently promotes it.
Selective insulin resistance in vascular endothelial cellsIn addition to vasodilator actions mainly via NO synthesis, insulin also has opposing vasoconstrictor actions to stimulate endothelin-1 (ET-1) production using mitogen-activated protein kinase (MAPK)-dependent (but not PI3K-dependent) signaling pathways in the endothelium [32-34]. ET-1 is a potent vasoconstrictor peptide synthesized and secreted from vascular endothelium that contributes to endothelial dysfunction and hypertension [34, 35]. Of note, in insulin-resistance state, selective inhibition of the IRS/PI3K/Akt pathway, but not MAPK cascade, is developed in vascular endothelial cells [33]. The pathway-selective impairment in PI3K-dependent insulin signaling in the vasculature, called “selective insulin resistance,” may contribute to reciprocal relationships between insulin resistance, endothelial dysfunction, and cardiovascular diseases.
Insulin signaling in vascular smooth muscle cellsThe effects of insulin on vascular smooth muscle cells (VSMCs) include p21 rat sarcoma virus (p21Ras) activation [36, 37], and mitogen-activated protein (MAP) kinase activation [38, 39] have already been reported. Although these proliferative effects are insulin concentration-dependent, no significant effects on MAP kinase activation or DNA synthesis promotion were observed when cultured smooth muscle cells were exposed to physiological concentrations of insulin [38, 40].
Because insulin activates both the IR and the insulin-like growth factor 1 receptor (IGF1R) at high concentrations, it is difficult to interpret whether IR or IGF1R mediates its effects. Both IR and IGF1R are expressed in VSMCs. In most vasculature cells, including VSMCs, IGF1R is approximately 8–10 times more abundant than IR, resulting in approximately 80% forming a hybrid IR/IGF1R form and 20% forming homo-IR. At physiological levels, insulin (1–10 nM) can only bind to homo-IR, while physiological levels of insulin-like growth factor 1 (IGF1) can activate both homo-IGF1R and hybrid IR/IGF1R. Thus, it is speculated that IGF1R binds to IR to form hybrid IR/IGF1R, thereby reducing the level of homo-IR and suppressing insulin effects [41]. The pathological role of hybrid IR/IGF1R in VSMCs in the development of vascular complications has yet to be elucidated.
Insulin signaling in macrophagesIn insulin-resistant humans, the activity of IR in monocytes is attenuated [42] and insulin-resistant macrophages have a strong proinflammatory effect. However, there are conflicting reports that high insulin concentrations in mononuclear cells and macrophages, including lymphocytes, have both anti-inflammatory and pro-inflammatory effects. In the ob/ob mouse model of obese type 2 diabetes, insulin signaling is attenuated and lipid uptake is enhanced in intraperitoneal macrophages; CD36 expression is increased at the level of post-translational modifications; and CD36 is upregulated in macrophage-specific IR-deficient mice with LDL receptor-deficient background exerting atherosclerosis in lysozyme M bacteriophage cyclization recombinant (LysM-Cre) mice. Increased cholesterol uptake by macrophages in aortic plaque lesions has been reported to induce apoptosis and necrosis, and promote atherosclerosis [43]. These results suggest that macrophage insulin resistance promotes atherosclerosis independently of systemic insulin resistance. However, there are contradictory reports that insulin resistance suppresses atherosclerosis in macrophage-specific IR-deficient and ApoE-deficient mice generated using LysM-Cre mice [44]. Therefore, it is possible that attenuated insulin signaling in macrophages contributes to the suppression of atherosclerosis by suppressing interleukin (IL)-6 and IL-1β expression by LPS. Finally, the significance of macrophage insulin signaling in atherosclerosis is varied, including forkhead transcription factors (FoxOs), and their roles may not be consistent across all mouse models and stages of atherosclerosis.
It has been suggested that persistent hyperinsulinemia promotes arteriosclerosis, but detailed studies using animal models are lacking. The effect of continuous subcutaneous administration of insulin pellets on arteriosclerosis in ApoE-deficient mice fed a high-fat diet (HFD) has been reported [45]. In HFD-fed ApoE-deficient mice, continuous insulin administration decreased aortic VCAM-1 expression, increased eNOS phosphorylation, and suppressed arteriosclerosis, associated with decreased plasma triglyceride, cholesterol, and lipoprotein levels. High-fat diet ApoE-deficient mice became obese and insulin resistant, suggesting that exogenous insulin contributes to hyperinsulinemia in this model. As a therapeutic strategy for obesity and type 2 diabetes, overcoming insulin resistance by exogenous insulin and activation of intravascular insulin signaling are expected to contribute to suppression of arteriosclerosis. Insulin receptor heterozygous ApoE-deficient mice also exhibited more hyperinsulinemia than ApoE-deficient mice, but there were no differences in insulin sensitivity, serum lipid levels, or blood pressure in aorta, skeletal muscle, liver, or adipose tissue [46]. These results suggest that hyperinsulinemia alone does not affect arteriosclerosis, and that changes in insulin signaling activity play an important role in the onset and progression of arteriosclerosis.
FoxOs are transcription factors with a forkhead domain and constitute a subfamily of FoxO1, FoxO3a, FoxO4, and FoxO6. FoxO6 is expressed almost exclusively in the central nervous system. FoxO1, FoxO3a, and FoxO4 are expressed in the vascular endothelial cells, VSMCs, and macrophages. Various post-translational modifications regulate FoxOs activity as a transcription factor, and its regulation by phosphorylation of Akt, a serine/threonine kinase, has been widely studied. When growth factors such as insulin activate Akt, FoxOs is phosphorylated in the nucleus and translocated to the nuclear envelope, where it becomes inactive as a transcription factor. Moreover, hyperglycemia is also reported to directly increase FoxOs activity by decreasing phosphorylation in cultured human aortic [28] and umbilical vein endothelial cells [47].
FoxOs are a multifunctional protein that regulates cell growth, differentiation, apoptosis, and stress tolerance, and has a variety of pathophysiological and physiological implications at the individual level. For example, in the liver, FoxO actively regulates gene expression of G6Pase (G6pc) and PEPCK (Pck1), which promotes glycolysis [48-51]. In addition, Notch signaling is also involved in the regulation of G6pc expression [52].
FoxOs in endothelial cellsInduction of atherosclerosis in LDL receptor-deficient mice has been reported to induce insulin resistance in large vessels and weaken FoxO1 and 3a phosphorylation, suggesting that FoxOs are activated in atherosclerotic vessels [53]. In vascular endothelial cells, FoxO1 and 3a positively regulate the transcriptional activity of inducible NOS (iNOS and Nos2) and negatively regulate eNOS (Nos3) [24, 28]. Vascular endothelial cell-specific deletions of FoxO1, 3a, and 4, using Tie2 promoter, markedly suppressed the development of atherosclerosis in LDL receptor-deficient mice [53]. In vascular endothelial cells of these mice, eNOS-derived nitric oxide (NO) production was increased and iNOS expression was decreased. Nuclear factor kappa B (NF-κB) activity, oxidative stress production, cellular senescence, and apoptosis were suppressed. Furthermore, the adhesion factors intercellular adhesion molecule-1 and VCAM-1 are target genes of FoxO1, indicating that FoxOs are an important integrative regulator of multiple mechanisms of atherosclerosis.
Hyperglycemia deacetylates FoxO1 in vascular endothelial cells and promotes its nuclear translocation [28]. Knock-in mice with mutants that constitutively deacetylate FoxO1 exhibited accelerated atherosclerosis through a myeloid cell-independent mechanism [54]. Insulin resistance and hyperglycemia, the main pathogenesis of type 2 diabetes, can then promote arteriosclerosis through the activation of FoxOs in vascular endothelial cells. However, it is unclear whether the target genes of dephosphorylated FoxOs activated by insulin resistance and the deacetylated FoxOs activated by hyperglycemia are the same. This interesting theme should be further investigated to elucidate the significance of insulin resistance and hyperglycemia in the onset and progression of arteriosclerosis in type 2 diabetes.
FoxO1 is also reported to produce metabolite S-2-hydroxyglutarate (S-2HG) in endothelial cells by inhibiting the mitochondrial enzyme 2-oxoglutarate dehydrogenase. Treatment of endothelial cells with 3-methyl-2-oxovalerate, a branched-chain amino acid catabolite, elicits S-2HG production and suppresses proliferation, causing vascular rarefaction in mice [55].
FoxOs in VSMCsApoptosis of VSMCs occurs at low levels in atherosclerotic plaques and in vascular remodeling. Akt is a major regulator of VSMC survival in vivo during vascular remodeling and atherogenesis, mediated in large part by the inhibition of FoxOs and its downstream genes. Plaque intimal VSMCs exhibit reduced p-Akt and a concomitant increase in unphosphorylated active FoxO3a [56]. FoxO3a promotes VSMC growth arrest and apoptosis after vascular injury [57, 58], suggesting that Akt-dependent regulation of FoxOs activity also plays an important role in VSMC biology. Moreover, in VSMCs of human pulmonary arteries, degradation of FOXO3 is promoted by interleukin-6-induced mammalian Ste20-like kinases 1/2 expression, which enhances proliferation and inhibits apoptosis in VSMCs to contribute to the pathogenesis of pulmonary arterial hypertension [59].
FoxOs in macrophagesFoxOs activation with insulin resistance was also observed in macrophages of obese and atherosclerotic mice [60]. However, atherosclerosis is promoted in systemic FoxO4-deficient mice [61] and in mice in which the bone marrow cell-specific FoxO1/3a/4 is knocked out by LysM-Cre [62]. One possible mechanism is that myeloid cell-specific FoxO1/3a/4-deficient mice may have increased myeloid cell division due to the presence of guanosine monophosphate (GMP), leading to neutrophil and monocyte hyperplasia with splenomegaly in the bone marrow. In addition, the percentage of Ly6Chi monocytes, a subset of atherosclerosis-promoting cells, was increased in peripheral blood. FoxOs in bone marrow cells acted as an inhibitor of atherosclerosis by modulating the number and expression of neutrophils and monocytes. In addition, LDL receptor deficiency was observed in bone marrow cells. Insulin resistance associated with increased nitrosylation/nitration of IR was deteriorated in the liver of mice lacking bone marrow cell-specific FoxOs in the background of LDL receptor deficient mice. This suggests that NO derived from inflammatory macrophages accumulated in the liver may induce nitrosylation/nitration of the IR receptor to promote insulin resistance in hepatocytes.
Thus, although FoxOs are activated in both vascular endothelial cells and macrophages in animal models of insulin resistance, interestingly enough, the pathophysiological importance of FoxOs in atherosclerosis is quite different. FoxOs may play a role in maintaining balance at the individual level in the pathogenesis of atherosclerosis by exerting cell- and organ-specific physiological effects (Fig. 1).
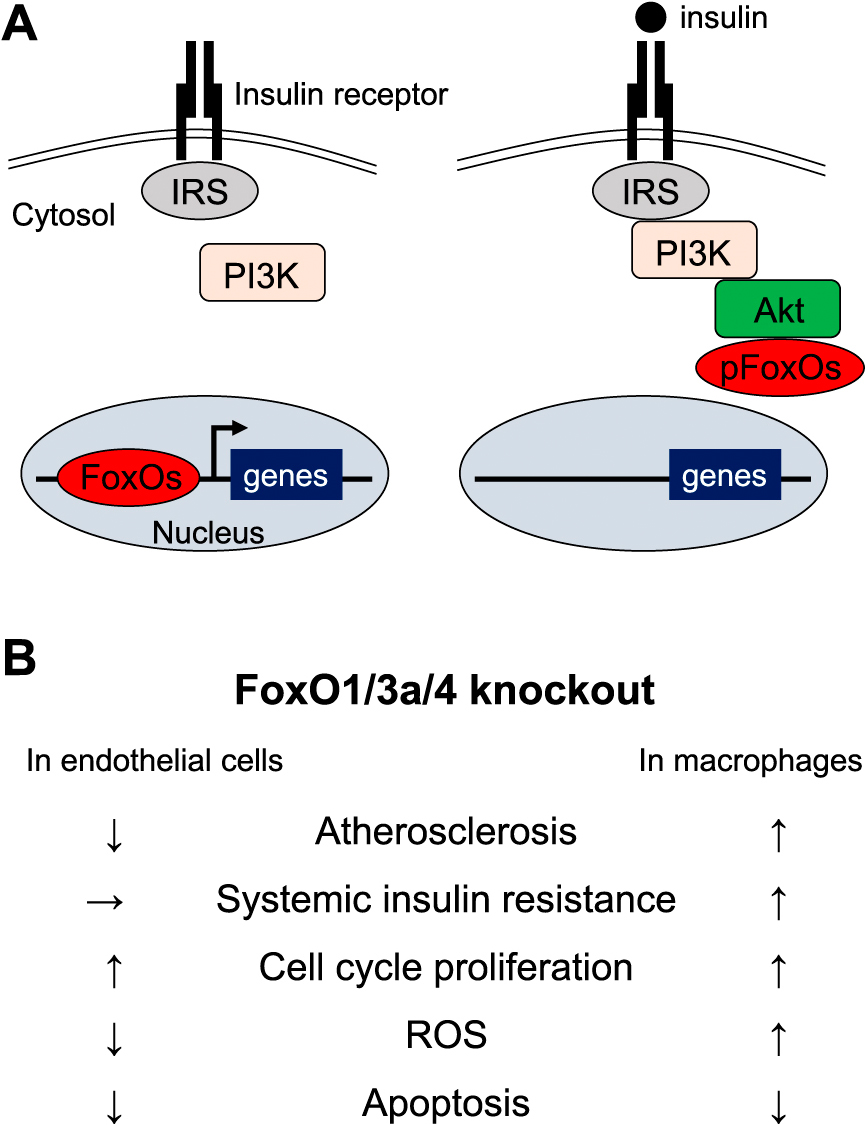
Regulation of FoxOs by insulin signaling and phenotypes of FoxOs-knockout mice.
A. Left: In the absence of insulin binding, FoxOs are transcriptional activator of multiple target genes. Right: Insulin binding to its receptor on the cell surface initiates PI3K and Akt activation, followed by FoxO phosphorylation, nuclear exclusion and loss of transcriptional activation. IRS, insulin receptor substrate; PI3K, phosphoinositide 3-kinases.
B. Summary of mice phenotypes in myeloid and endothelial cells-specific FoxO1/3a/4 knockout mice. ROS, reactive oxygen species; iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase.
PA is secondary hypertension characterized by autonomous aldosterone hypersecretion due to adrenocortical adenoma and/or hyperplasia. Recently, accumulating lines of evidence reveal its higher prevalence (about 5–15%) among unselected hypertensive patients than previously thought [63-65]. In addition, several clinical studies have shown that patients with PA have a higher incidence of proteinuria, cerebral hemorrhage, and left ventricular hypertrophy than age- and sex-matched control patients with essential hypertension despite the higher blood pressures and longer durations in control patients [66-69]. These results suggest that aldosterone is directly involved in the development of cardiovascular injury by mechanism(s) other than its sodium-water retention and hypertensive effect (Fig. 2).
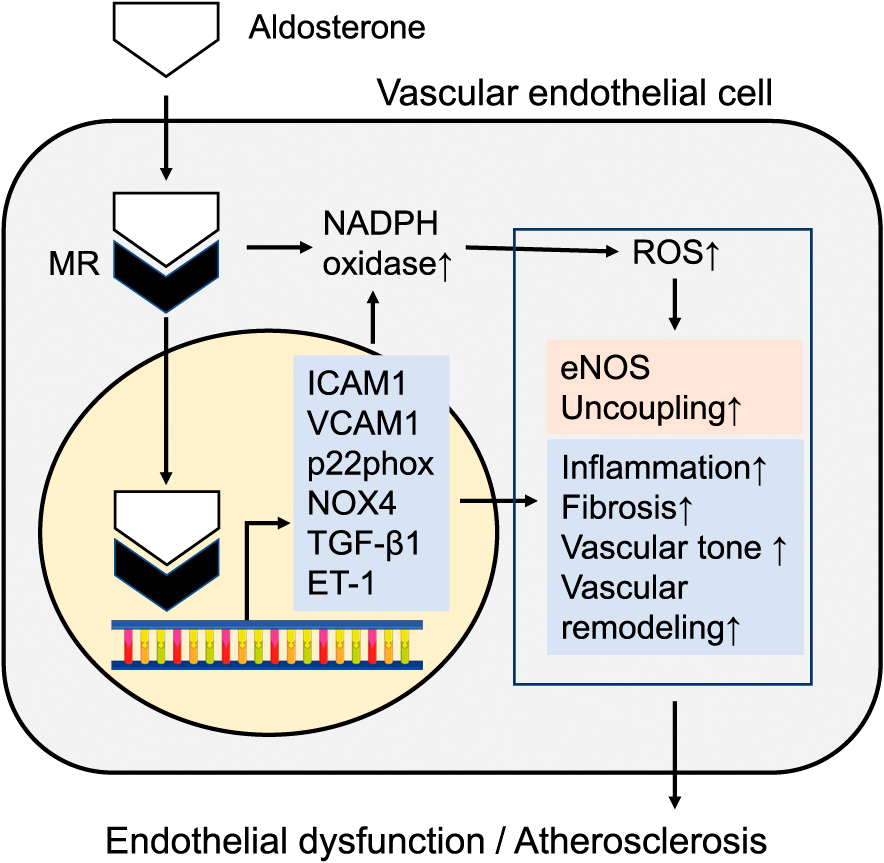
Aldosterone actions in vascular endothelial cells.
Mineralocorticoid receptor activation by aldosterone in endothelial cells can contribute to amplify cardiovascular adverse outcomes through genomic and non-genomic actions, exacerbating vascular tone through the induction of augmented reactive oxygen species (ROS) production, vascular inflammation, fibrosis, and remodeling, finally leading to endothelial dysfunction and atherosclerosis. eNOS, endothelial nitric oxide synthase; NOX4, NADPH Oxidase 4; ICAM1, Intercellular Adhesion Molecule 1; VCAM1, vascular cell adhesion molecule 1; TGF-β1, transforming growth factor beta 1
Patients with CS also have a higher mortality rate of cardiovascular complications (myocardial infarction, congestive heart failure, stroke) than the age- and gender-matched normal population [70]. Excess glucocorticoids also directly affect the cardiovascular system by affecting the renin-angiotensin system, the sympathetic nervous system, and the kallikrein-kinin system [71]. It has been suggested that glucocorticoid excess in CS plays an important role in developing cardiovascular complications.
Endothelial dysfunction in PA and CSEndothelial dysfunction is considered an initial event in the development of CVD [72]. Measurement of flow-mediated vasodilation (FMD) of the brachial artery by an ultrasound technique has been widely used as a reliable non-invasive method for evaluating endothelial function [73]. This technique is currently considered one of the standard tests for assessing conduit artery endothelial function and a potential predictor for cardiovascular events based on the results of many clinical trials.
The FMD in patients with PA was reported to be significantly lower than in the blood pressure-matched control subjects [74]. The FMD showed significant negative correlations with systolic blood pressure, brachial-ankle pulse wave velocity, plasma aldosterone concentration (PAC), and the aldosterone/renin ratio (ARR). The FMD significantly improved after surgical and medical treatment of aldosteronism, although the changes in FMD did not correlate with blood pressure, PAC, or ARR. In patients with CS, the FMD was also significantly lower than that in control subjects [75]. The FMD in patients with CS showed significant negative correlations with morning serum cortisol levels and 24-h urinary free cortisol excretion. Accordingly, endothelial dysfunction in patients with PA and CS may be related to hormonal excess rather than hypertension and can possibly be reversed with treatment.
Are aldosterone and cortisol cardiovascular risk hormones?Even in patients with hypertension, including subjects without being diagnosed as PA, FMD was negatively and significantly correlated with plasma aldosterone and 24-hour urinary aldosterone. However, ARR was independent of blood pressure, age, and body mass index [76]. Furthermore, elevated plasma aldosterone levels are seen in numerous clinical conditions, including hypertension and heart failure, with evidence suggesting that increased aldosterone levels are associated with greater CVD mortality [77, 78]. A prospective cohort study of 3,153 patients (average follow-up 7.7 years) in which plasma aldosterone levels were measured at the time of coronary angiography has been published. It suggested greater all-cause and cardiovascular mortality among patients in the top three quartiles of PAC (most of whom had an aldosterone concentration considered to be in the normal range for this assay) compared with those in the lowest quartile, even after adjusting for traditional coronary heart disease risk factors [78]. It has also been reported that unsuppressed plasma cortisol concentration (>1.8 μg/dL) after low-dose dexamethasone suppression tests in patients with incidental adrenocortical adenomas is associated with increased mortality, mainly related to CVD and infection [73]. Taken together, plasma aldosterone and cortisol concentrations seem to be associated with cardiovascular risks in subjects with or without PA or CS. Further studies are needed to evaluate the long-term cardiovascular prognosis in subjects with hyperaldosteronemia or hypercortisolemia.
As discussed above, systemic and local insulin resistance is a fundamental pathophysiology that promotes atherosclerosis, and the activation of insulin signaling in cells of vascular systems and macrophages may antagonize the condition. Although persistent hyperinsulinemia was initially thought to act to promote atherosclerosis, recent reports at the animal level suggest that insulin administration to insulin-resistant individuals may also inhibit atherosclerosis. Furthermore, endocrine disorders associated with cardiovascular risk should be appropriately screened and treated. It is hoped that findings from these basic research and clinical studies will lead to further discussion and research on the prevention of cardiovascular complications in endocrine metabolic diseases.
I would like to express my sincere gratitude to Professor Emeritus Yukio Hirata, Professor Emeritus Masayoshi Shichiri, Dr. Takanobu Yoshimoto, Professor Domenico Accili, Professor Yoshihiro Ogawa, and Former Professor Kenichiro Kitamura for their advice and encouragement of my research.
Kyoichiro Tsuchiya declares that he has no conflict of interest.
Statement of animal and/or human participantsThis article does not contain any studies with human or animal subjects.