Case Presentation
A 46-year-old female underwent emergency laparoscopic appendicectomy for an appendiceal perforation with localized peritonitis. The pathohistological diagnosis was goblet cell carcinoma of the appendix, and the patient underwent laparoscopic resection of the right colon. During surgery, occasional paroxysmal hypertension with a systolic blood pressure increase >200 mmHg was observed. She was referred to our division for further endocrine evaluation. The patient has been healthy since birth, and has never experienced symptoms such as headache, palpitations or hyperhidrosis that could be associated with PPGL. Moreover, no hypertension or glucose intolerance were noted, and no family history of hereditary endocrine disorders was documented; however, PPGL was suspected due to paroxysmal blood pressure elevation during surgery. Upon reviewing the patient’s medical records, we noted that she had a history of hepatic angiolipoma, which was present in the arterial phase in the computed tomography (CT) scan as a heterogeneously stained mass (55 mm in diameter) in the posterior right lobe of the liver (Fig. 1A). A more detailed analysis using multidetector-row CT failed to identify a normal right adrenal gland surrounding the tumor. In the screening of the catecholamine profile, all types of catecholamine levels, including normetanephrine/noradrenaline, metanephrine/adrenaline, and dopamine, were elevated (Table 1). Abdominal magnetic resonance imaging (MRI) revealed tumor heterogeneity and hyperintensity predominantly on T2-weighted images (Fig. 1B). 123I-metaiodobenzylguanidine (MIBG) single-photon emission computed tomography (SPECT) demonstrated accumulation in the tumor with no other significant accumulation (Fig. 1C). A clinical diagnosis of pheochromocytoma rather than paraganglioma was made based on the pattern of catecholamine elevation and tumor localization on imaging studies (Table 1).

Table 1
Complete blood count, biochemistry data, and catecholamine profile at admission
| Parameter |
Value |
Reference range |
| White-cell count (/μL) |
3,600 |
4,500–8,000 |
| Neutrophils (%) |
63.1 |
40–70 |
| Eosinophils (%) |
8.8 |
0.0–7.0 |
| Basophils (%) |
1.4 |
0.0–1.0 |
| Monocytes (%) |
5.8 |
2.0–7.0 |
| Lymphocytes (%) |
20.9 |
27–47 |
| Hemoglobin (g/dL) |
9.8 |
12–16 |
| Platelet count (/μL) |
356,000 |
100,000–330,000 |
| Total protein (g/dL) |
7.2 |
6.3–8.2 |
| Albumin (g/dL) |
4.7 |
3.5–5.0 |
| Urea nitrogen (mg/dL) |
13.5 |
8.0–20.0 |
| Creatinine (mg/dL) |
0.66 |
0.5–1.2 |
| Sodium (mEq/L) |
138 |
135–147 |
| Potassium (mEq/L) |
4.4 |
3.3–4.8 |
| Chloride (mEq/L) |
106 |
98–108 |
| Aspartate aminotransferase (U/L) |
20 |
8–38 |
| Alanine aminotransferase (U/L) |
13 |
4–44 |
| Lactate dehydrogenase (U/L) |
196 |
106–211 |
| Alkaline phosphatase (U/L) |
50 |
104–338 |
| γ-glutamyl transpeptidase (U/L) |
12 |
16–73 |
| Creatine kinase (U/L) |
80 |
25–170 |
| C-reactive protein (mg/dL) |
0.01 |
0.0–0.3 |
| Glucose (mg/dL) |
69 |
73–109 |
| HbA1c (%) |
5.8 |
4.9–6.0 |
| PT-INR |
1.00 |
0.85–1.15 |
| APTT (sec) |
27.1 |
26–38 |
| D-dimer (μg/mL) |
0.5 |
<1.0 |
| free metanephrine (pg/mL) |
233 |
0–130 |
| free normetanephrine (pg/mL) |
1,670 |
0–506 |
| Urine |
| total metanephrine (mg/day) |
1.0 |
0.05–0.2 |
| total normetanephrine (mg/day) |
3.2 |
0.1–0.28 |
| adrenalin (μg/day) |
40.4 |
1.1–22.5 |
| noradrenalin (μg/day) |
410 |
29.2–118 |
| dopamine (μg/day) |
1,500 |
100–1,000 |
| VMA (mg/day) |
20.6 |
1.4–4.9 |
HbA1c: hemoglobin A1c, PT-INR: prothrombin time/international normalized ratio, APTT: Activated partial thromboplastin time, VMA: vanillylmandelic acid
Based on the diagnostic guidelines for PPGL, doxazosin treatment was initiated and titrated up to 12 mg. Metyrosine was administered and titrated up to 750 mg; consequently, both plasma metanephrine and normetanephrine levels were reduced by approximately half (Table 2). Tumor resection was performed laparoscopically, and the catecholamine and metabolite levels were subsequently normalized (Table 2).
Table 2
Catecholamine profile alternation before initiation of medication, post-medication and post-surgery
| Parameter |
Pre-medication |
Post-medication |
Post-surgery |
Reference range |
| free metanephrine (pg/mL) |
233 |
152 |
21 |
0–130 |
| free normetanephrine (pg/mL) |
1,670 |
789 |
48 |
0–506 |
| Urine |
| total metanephrine (mg/day) |
1.0 |
|
0.10 |
0.05–0.2 |
| total normetanephrine (mg/day) |
3.2 |
|
0.15 |
0.1–0.28 |
| adrenalin (μg/day) |
40.4 |
|
5.6 |
1.1–22.5 |
| noradrenalin (μg/day) |
410 |
|
63.4 |
29.2–118 |
| dopamine (μg/day) |
1,500 |
|
480 |
100–1,000 |
| VMA (mg/day) |
20.6 |
|
3.2 |
1.4–4.9 |
VMA: vanillylmandelic acid
The resected tumor was 55 mm in diameter. Grossly, the tumor was solid with a mixture of dark and pale-gray areas. The adrenal gland, compressed by the tumor, retained its normal structure, whereas the cortex and medulla were preserved. The tumor and normal adrenal gland were in close proximity but not contiguous, indicating a paraganglioma rather than a pheochromocytoma of ectopic adrenal origin (Fig. 2A). Interestingly, the tumors exhibited two distinct histological patterns. Both components are relatively well demarcated. The tumor was located outside the normal right adrenal gland and was separated by a capsule (Fig. 2B). Most of the tumor (70%) was composed of polygonal cells with round nuclei and amphophilic broad cytoplasm, mainly in a chordate to focal formation against a background of vascular fibrocardium, with a Zellballen structure (Fig. 2C), indicating a paraganglioma. Adjacent to the paraganglioma, mature ganglion cells and intervening Schwann cell-rich stroma were observed surrounding the margins of the tumor, indicating a ganglioneuroma (Fig. 2D). Based on these histological findings, the tumor was diagnosed as a composite paraganglioma consisting of a paraganglioma and ganglioneuroma. Immunohistochemical examination revealed that the paraganglioma cells and some ganglion cells in the ganglioneuroma were positive for chromogranin A (Fig. 2E, F). S100 was positive in both paraganglioma sustentacular and ganglioneuroma Schwann cells (Fig. 2G, H).

The Grading System for Adrenal Pheochromocytoma and Paraganglioma (GAPP) score was 3 points (intermediate cellularity, 1 point; capsular invasion, 1 point; catecholamine type, norepinephrine type, 1 point), which was within the category of moderate differentiation. The composite pheochromocytoma/paraganglioma prognostic score was 0, indicating a low risk of metastasis and progression of PPGLs [5].
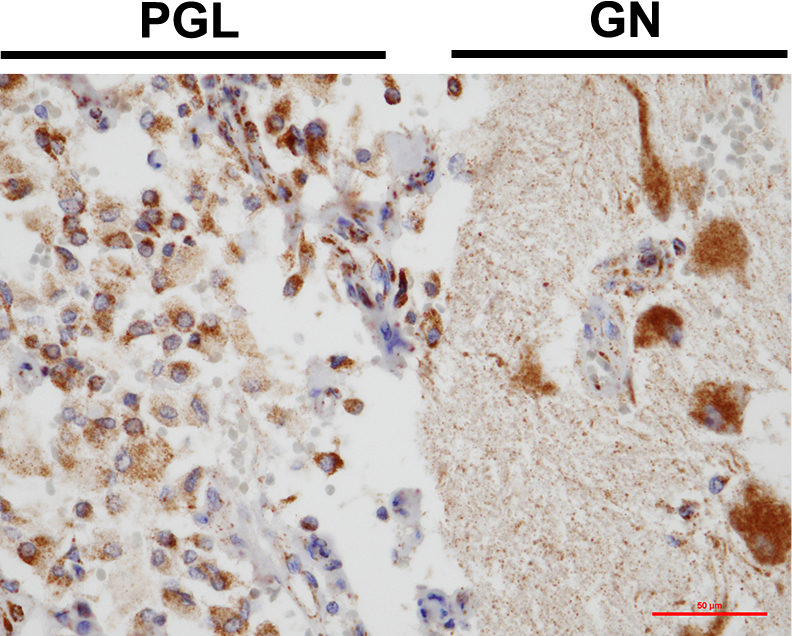
To address the pathological discrepancy in which both normetanephrine and metanephrine levels were elevated despite the diagnosis of paraganglioma, we performed immunostaining analysis to confirm whether PNMT was expressed in tumor cells. Interestingly, PNMT was focally positive in the paraganglioma and ganglion cells of the ganglioneuroma (Fig. 3A, B), whereas the normal ganglion (Fig. 3C) and paraganglia (Fig. 3D) were weakly positive for PNMT. On the other hand, strong positive PNMT expression was observed in the normal right adrenal medulla adjacent to the tumor (Fig. 3E). To elucidate the detailed pathogenesis, we investigated the SDHB expression in tumor tissues by immunohistochemistry, which exhibited that SDHB was well-conserved in both paraganglioma and ganglioneuroma (Fig. 4).

Although catecholamine levels decreased to normal after surgery, hypertension persisted, and treatment with 5 mg amlodipine was initiated. Three years after surgery, no evidence of recurrence or metastasis has been observed [6].
Discussion
A composite PPGL is a tumor consisting of two or more components of neuronal origin in addition to a chromaffin cell tumor. Pheochromocytomas are known to form composite tumors consisting of ganglioneuromas (61%), ganglioblastomas (16%), neuroblastomas (11%), and schwannomas (1%) [7]. Ganglioneuroma is a benign neurogenic tumor that originates from the sympathetic ganglion. This tumor was recently considered a mature differentiated neuroblastoma [8]. Various sites are associated with the presence of sympathetic ganglions, including the posterior mediastinum and retroperitoneum, which account for approximately 40%, and a further 20–30% arise from the adrenal medulla [7]. Ganglioneuromas are diagnosed more often in females and in those aged <20 years [7]. Although they are generally silent both clinically and endocrinologically, several cases have been reported of functioning tumors that secrete various hormones, such as catecholamines and their metabolites, ACTH (rarely), cortisol, androgens, and vasoactive intestinal polypeptides [9].
However, little is known about the pathogenesis of composite PPGLs. Because chromophilic cells differentiate from sympathetic nerve matrix cells, they are thought to be generated during tumorigenesis at an early stage of differentiation [9]. Copy number aberrations in the MYCN gene is considered a major oncogenic driver of neuroblastoma [10]. However, the amplification thereof has not been identified in the neuroblastic component of composite pheochromocytomas [11]. In contrast, BRAF and HRAS mutations, which are associated with PPGLs pathogenesis, are more frequently identified in composite paragangliomas than SDH or ATRX [3]. These results suggest that the tumorigenesis of neuroblastoma/ganglioneuroma components in composite PPGLs may have a different mechanism from that of neuroblastoma in general; that is, transdifferentiation from the PPGLs component. In addition, it has recently been reported that mutations in NF1, RET, VHL, and MAX genes, the driver genes of germline PPGLs, are present in composite PPGLs, suggesting that the pathogenesis of PPGLs may also be implicated in the pathogenesis of composite tumors [12-16].
To the best of our knowledge, composite paragangliomas are extremely rare, with only 33 cases reported to date, excluding our present case. The characteristics of all cases are summarized in Table 3. The ages of the patients ranged from 13 to 81 years, and 17 of the 32 patients were women. Retroperitoneal origin was the most common tumor localization, accounting for half of all cases, followed by bladder and seminal cord origins. The diameters of composite paragangliomas ranged from 1.0 to 11.0 cm. In 28 of the 33 cases, paragangliomas coexisted with a ganglioneuroma component, the other three cases with neuroblastoma, and two cases with a ganglioneuroblastoma component. Although patients with composite pheochromocytomas and paragangliomas share similar characteristics such as age, sex, and tumor size, ganglioneuroma is more common in those with paragangliomas in terms of composite tumor component histology, as in this case [7]. Two cases of NF1, one of MEN2B, and one of SDHB have been reported in relation to a genetic disease with germline mutations. Moreover, an SDHB mutation of the tumor was detected in one of the patients.
Table 3
Summary of 34 patients with composite paraganglioma
| Year [ref] |
Age |
Sex (M/F) |
Tumor location |
Size (cm) |
Composite type |
Elevated catecholamine type |
Genetic abnormalities |
| 1972 [33] |
29 |
M |
Filum terminale |
2.0 × 1.2 |
PA-GN |
|
|
| 1978 [34] |
65 |
F |
Duodenum |
1.0 |
PA-GN |
|
|
| 1980 [35] |
49 |
F |
Bladder |
3.5 |
PA-GN |
M, V |
|
| 1982 [36] |
33 |
F |
Conus medullaris region |
4 × 2.5 × 2.5 |
PA-GN |
|
|
| 1989 [37] |
48 |
M |
Organ of Zuckerkandl |
NA |
PA-GN |
|
|
| 1991 [38] |
21 |
F |
Retroperitoneal |
6.2 × 5.6 |
PA-GNB |
NA, D |
|
| 1991 [38] |
68 |
M |
Retroperitoneal |
7.0 × 7.0 × 6.0 |
PA-GN |
NA |
|
| 1992 [39] |
48 |
M |
Retroperitoneal |
7.0 × 8.0 × 5.0 |
PA-GN |
NA, A, D |
|
| 1998 [40] |
81 |
F |
Bladder |
4.8 |
PA-GN |
|
|
| 2003 [41] |
70 |
F |
Bladder |
6.5 |
PA-GN |
NA, A |
|
| 2005 [42] |
70 |
M |
Posterior mediastinum |
4.0 × 3.5 × 2.5 |
PA-GN |
|
|
| 2005 [43] |
73 |
M |
Bladder |
4.0 × 3.0 × 2.5 |
PA-GN |
NA, D, V |
|
| 2005 [44] |
74 |
F |
Nerve roots of the cauda equina dorsally |
1.8 |
PA-GN |
|
|
| 2006 [45] |
NA |
F |
Retroperitoneal (pancreas) |
NA |
PA-GN |
|
|
| 2009 [46] |
13 |
F |
Retroperitoneal |
NA |
PA-NB |
|
SDHB |
| 2009 [47] |
57 |
F |
Retroperitoneal (pancreas) |
3.2 × 2.5 |
PA-GN |
|
|
| 2009 [47] |
63 |
F |
Retroperitoneal |
6.5 × 5.0 × 3.0 |
PA-GN |
|
|
| 2009 [48] |
64 |
M |
Bladder |
5.0 |
PA-GN |
|
|
| 2010 [49] |
31 |
F |
Retroperitoneal |
6.0 × 5.0 × 4.6 |
PA-GN |
|
|
| 2010 [50] |
47 |
M |
Filum terminale |
2.6 × 1.7 × 1.2 |
PA-GN |
|
|
| 2010 [51] |
50 |
M |
Retroperitoneal |
4.5 × 4.0 × 2.5 |
PA-GN |
|
|
| 2010 [52] |
61 |
M |
Retroperitoneal |
11.0 |
PA-NB |
NM |
|
| 2012 [53] |
34 |
F |
Retroperitoneal |
6.8 × 4.9 × 4.6 |
PA-GN |
|
NF1 |
| 2012 [54] |
68 |
M |
Retroperitoneal |
3.0 × 2.2 × 2.0 |
PA-GN |
|
|
| 2013 [55] |
51 |
NA |
Intraspinal extradural lesions |
NA |
PA-GNB |
|
|
| 2013 [4] |
52 |
F |
Retroperitoneal |
6.0 × 5.0 × 4.0 |
PA-GN |
|
|
| 2015 [56] |
44 |
M |
Intraosseous sacral lesion with invasion of sacrum |
NA |
PA-GN |
|
|
| 2015 [57] |
45 |
M |
Bladder |
4.4 |
PA-NB |
|
|
| 2017 [58] |
55 |
F |
Bladder |
6.2 × 5.9 |
PA-GN |
|
|
| 2017 [59] |
59 |
M |
Retroperitoneal |
3.0 |
PA-GN |
M, V |
MEN2B |
| 2017 [60] |
67 |
M |
Retroperitoneal |
6.2 |
PA-GN |
NA/NM, D |
NF1 |
| 2019 [61] |
50 |
F |
Right carotid space |
5.9 × 1.2 × 0.5 |
PA-GN |
|
SDHB* |
| 2022 [62] |
64 |
F |
Retroperitoneal (celiac trunk) |
6.0 × 4.2 × 3.7 |
PA-GN |
|
|
| Present |
46 |
F |
Retroperitoneal |
5.5 |
PA-GN |
NA/NM, A/M, D |
|
PA: paraganglioma, GN: ganglioneuroma, GNB: ganglioneuroblastoma, NB: neuroblastoma, M: metanephrine, NM: normetanephrine, NA: noradrenaline, A: adrenaline, D: dopamine, V: VMA
* somatic mutation
Of the 33 cases reported, catecholamines were measured in only nine cases; of which, seven had elevated noradrenaline/normetanephrine, four had elevated adrenaline/metanephrine, two had elevated levels of both, four had elevated dopamine, and only one exhibited elevated levels of all catecholamines, as in the present case. However, the urinary dopamine level in our case was only 1.5 times higher than the normal range, and no symptoms associated with dopamine excess, such as fever, malaise, weight loss, or diarrhea, were observed [17].
Paragangliomas tend to secrete noradrenaline more frequently than adrenaline, with approximately 80–90% of paragangliomas secreting predominantly noradrenaline and only 10–20% secreting a combination of noradrenaline and adrenaline [18, 19]. In contrast, noradrenaline is elevated in most cases of pheochromocytoma, many of which secrete both adrenaline and noradrenaline, and cases in which only adrenaline is elevated are rather rare [20].
Because PNMT is a critical molecule for adrenaline synthesis in the adrenal medulla, we also examined its expression in the current case [21]. PNMT converts noradrenaline to adrenaline by rearranging the methyl groups of S-adenosylmethionine. PNMT is primarily expressed in the adrenal medulla, heart, brainstem, muscles, and kidneys [22]. According to our immunohistochemical analysis, both paraganglioma and ganglioneuroma cells were focally positive for PNMT, whereas normal ganglia and paraganglia were weakly expressed in non-tumoral samples.
Although glucocorticoids (GC) induce PNMT expression in the adrenal medulla, previous studies reported that sufficient PNMT transcription was not induced in in vivo experiments in which pheochromocytoma and normal adrenal cortex were in direct contact or primary cultured pheochromocytoma cells were treated with dexamethasone [23-25]. In addition, the presence of a capsule between the normal adrenal gland and tumor in this case and in previously reported case where PNMT expression was confirmed in a paraganglioma originating from the heart, far from the adrenal gland, diminished the likelihood the that PNMT expression in the paraganglioma is a result of paracrine secretion of GCs from the normal adrenal gland [23].
In addition to GC/GC receptor, several other transcription factors, including early growth response gene-1, specificity protein 1, glial cell missing-like factor, activating protein 2, and c-Myc-activating factor are involved in activating PNMT transcription [26]. PNMT hypermethylation has also been reported to be involved in the synthetic downregulation of SDH- and VHL-related tumors [27, 28]. Taken together, the high GC environment near the adrenal glands may affect PNMT expression. Further investigation is needed to confirm whether other factors also influence PNMT expression.
Although a positive correlation between intratumoral PNMT expression levels and adrenaline synthesis has been shown in pheochromocytomas, its expression in paragangliomas varies (ranging from 8% to 75%), regardless of the catecholamine profile [29, 30]. Scoring to quantify PNMT using immunostaining failed to distinguish between pheochromocytomas and paragangliomas [31]. In our case, PNMT was strongly expressed in the compressed normal right adrenal medulla. Moreover, strong PNMT expression was observed in the tumor, especially in the paraganglioma region; however, only weak expression was observed in the normal ganglion and the normal paraganglia. Thus, PNMT expression in the tumor may have modified the catecholamine profile. Although it remains unclear how PNMT is expressed in paraganglioma and ganglioneuroma tumors, mutations in BRAF, HRAS, NF1, RET, VHL and MAX may be involved.
However, in the present case, there were no clinical findings suggestive of hereditary PPGL, such as medullary thyroid carcinoma, café-au-lait spots, or family history, and SDHB expression in the tumor was preserved. Four cases of composite paraganglioma were examined for SDHB mutations, including the present case, and two of them showed mutations, whereas none of the seven cases of composite pheochromocytoma had SDHB mutations (Table 3). However, since the frequency of SDHB mutations is known to be higher in abdominal paragangliomas than in pheochromocytomas, we cannot conclude whether SDHB mutations contribute to the pathogenesis of composite paragangliomas; further case analyses are needed [32]. In addition, our case report lacks adequate genetic background studies. This prevented us from excluding all previously reported genetic variants associated with the combined form of PPGL and paraganglioma-ganglioneuroma.
In summary, we present a rare case of composite paraganglioma with a pheochromocytoma-like catecholamine profile that expressed PNMT in both the paraganglioma and ganglioneuroma component cells.