Abstract
We report three Japanese patients with Sotos syndrome accompanied by marked overgrowth, i.e., a 2 8/12-year-old boy with a height of 105.2 cm (+4.4 SD) (patient 1), the mother of patient 1 with a height of 180.8 cm (+4.1 SD) (patient 2), and a 12 10/12-year-old girl with a height of 189.4 cm (+6.3 SD) (patient 3). In addition to the marked overgrowth (tall stature), patients 1–3 exhibited Sotos syndrome-compatible macrocephaly and characteristic features, whereas intellectual and developmental disabilities remained at a borderline level in patient 1 and were apparently absent from patients 2 and 3. Thus, whole exome sequencing was performed to confirm the diagnosis, revealing a likely pathogenic c.6356A>G:p.(Asp2119Gly) variant in NSD1 of patients 1 and 2, and a likely pathogenic c.6599dupT:p.(Ser2201Valfs*4) variant in NSD1 of patient 3 (NM_022455.5). The results, in conjunction with the previously reported data in nine patients with marked overgrowth (≥4.0 SD), imply that several patients with Sotos syndrome have extreme tall stature even in adulthood. Thus, it is recommended to examine NSD1 in patients with marked overgrowth as the salient feature.
SOTOS SYNDROME (OMIM #117550) is a rare congenital malformation syndrome caused by heterozygous loss-of-function sequence variants or microdeletions affecting NSD1 on chromosome 5q35 [1, 2]. It is associated with overgrowth (tall stature) with advanced bone age, macrocephaly, characteristic features, and intellectual and developmental disabilities (IDDs) [1]. Overgrowth is salient in infancy to toddlerhood, although the adult height usually remains at the upper-normal range, primarily because of advanced bone age and resultant premature epiphyseal fusion [1, 3]. Macrocephaly is also conspicuous in infancy to toddlerhood, and appears to be recognizable at all ages [1]. Characteristic features are pathognomonic and most evident in early childhood. IDDs are manifested by most patients, although they are associated with incomplete penetrance and variable expressivity. In addition, this syndrome is also often accompanied by cardiac anomalies, renal anomalies, scoliosis, and seizure [1].
Here, we report three Japanese patients who had Sotos syndrome with marked overgrowth and likely pathogenic NSD1 variants, and review similar cases reported in the literature. The results imply that NSD1 should be examined in patients with extreme tall stature as the salient feature.
Case Reports
Patient 1
This Japanese boy was born to non-consanguineous parents 41 weeks of gestation after an uncomplicated pregnancy and delivery. The mother was extremely tall (patient 2), whereas the father was of normal height. At birth, his length was 62 cm (+7.0 SD), his weight 5.56 kg (+5.6 SD), and his occipitofrontal circumference (OFC) 40 cm (+5.1 SD). After birth, he manifested somewhat delayed psychomotor development: he controlled his head at five months of age, stood with support at one year of age, and walked without support at one year and seven months of age. At two years and eight months of age, he was referred to us because of excessive growth (Fig. 1A). His height was 105.2 cm (+4.4 SD), his weight 21.8 kg (+6.5 SD), and his OFC 55.0 cm (+4.3 SD). Bone survey indicated advanced bone age and otherwise normal skeletal findings (Fig. 1B, Table 1). His developmental quotient was evaluated as 78 by the new version of the k-type developmental test. Physical examination showed multiple facial features, and endocrine studies revealed low serum IGF-I (Table 1). Ultrasound studies showed no cardiac or renal malformation.

Table 1
Clinical features of patients 1–3
| Age at examination |
Patient 1
2 yrs 8 mos |
Patient 2
(Mother of patient 1)
Adult |
Patient 3
12 yrs 10 mos |
| Physical findings |
| Height (cm, SDS) |
105.2 (+4.4) |
180.8 (+4.1) |
189.4 (+6.3) |
| Overgrowth (Tall stature)* |
+ |
+ |
+ |
| OFC (cm, SDS) |
55.0 (+4.3) |
NE |
63.8 (+5.6) |
| Macrocephaly* |
+ |
+† |
+ |
| Advanced bone age |
+ (3 y 7 m) |
Unknown |
+ (14 y 1 m) |
| IDDs |
+ (borderline) |
– |
– |
| Facial gestalt |
| Coarse appearance |
+ |
+ |
– |
| Prominent forehead |
+ |
+ |
+ |
| Hypertelorism |
+ |
+ |
– |
| Downward slanting fissures |
+ |
+ |
– |
| Large ears |
+ |
+ |
+ |
| Pointed chin |
+ |
+ |
+ |
| Cardiac abnormality |
– |
– |
– |
| Renal abnormality |
– |
– |
– |
| Scoliosis |
– |
– |
– |
| Endocrine data |
| IGF-I (ng/mL) |
6 (18–154) |
NE |
226 (188–654) |
| GH (ng/mL) |
2.98 (<6) |
NE |
0.44 (<6) |
| Intact PTH (pg/mL) |
NE |
NE |
51.1 (10–65) |
| TSH (μIU/mL) |
1.4 (0.6–4.2) |
NE |
3.7 (0.6–4.2) |
| Free T3 (pg/mL) |
NE |
NE |
3.1 (2.5–4.1) |
| Free T4 (ng/dL) |
1.4 (0.8–1.5) |
NE |
1.1 (0.8–1.5) |
| ACTH (pg/mL) |
NE |
NE |
14.7 (7.2–63) |
| Cortisol (μg/dL) |
8.3 (7.1–19.6) |
NE |
4.5 (7.1–19.6) |
| LH (mIU/mL) |
NE |
NE |
2.5 (1.1–14.2) |
| FSH (mIU/mL) |
NE |
NE |
3.8 (1.5–8.5) |
| E2 (pg/mL) |
NE |
NE |
115 (44–491) |
Abbreviation: OFC, occipitofrontal circumference; IDDs, intellectual and developmental disabilities; and NE, not examined.
The values in parentheses represent the age- and sex-matched reference values.
* ≥+2.0 SD
† Clinical assessment.
This patient was the mother of patient 1. She had marked tall stature of 180.8 cm (+4.1 SD). Although the assessment of clinical findings remained quite fragmentary, she had Sotos syndrome-compatible phenotype such as apparent macrocephaly and facial features but was apparently free from IDDs (Table 1).
Patient 3
This Japanese girl was born at term to non-consanguineous and healthy parents with normal heights. At birth, her length was 54 cm (+2.7 SD), and her weight 4.1 kg (+3.2 SD). Except for overgrowth, her postnatal clinical course was uneventful, without discernible IDDs and cardiac and renal anomalies. At 12 years and 10 months of age, she was referred to us because of excessive tall stature (Fig. 1C). Her height was 189.4 cm (+6.3 SD), her weight 87.5 kg (+4.8 SD), her arm span 199 cm (+6.9 SD), and her OFC 63.8 cm (+5.6 SD). Roentgenographic studies indicated advanced bone age and no skeletal malformation (Fig. 1D, Table 1). Physical examination revealed several facial features, and endocrine studies showed a low-normal serum IGF-1 value (Table 1). Ultrasound studies showed no cardiac or renal malformation.
Molecular Studies
This study was approved by the Institutional Review Board Committee at Hamamatsu University School of Medicine, and was performed after obtaining written informed consent. While overgrowth, macrocephaly, and facial features suggested Sotos syndrome in patients 1–3, their heights were extremely tall (≥+4.0 SD) and they lacked obvious IDDs. Thus, we attempted to have a molecular diagnosis. Here, considering that overgrowth is a heterogeneous condition [4], we performed whole exome sequencing with SureSelect Human All Exon V6 (Agilent Technologies) using leukocyte gDNA samples from patient 1 and his parents (patient 2 and the father) and those from patient 3 and her mother. Captured libraries were sequenced using NextSeq 500 (Illumina) with 150 bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as described previously [5]. Human GRCh38 was utilized as the reference genome. We extracted rare variants with minor allele frequencies of ≤0.01 in all the public and in-house databases utilized in this study, and performed in silico pathogenicity predictions for extracted rare variants by several methods. The databases and pathogenicity prediction methods are described in the legend for Fig. 2, together with their URLs.
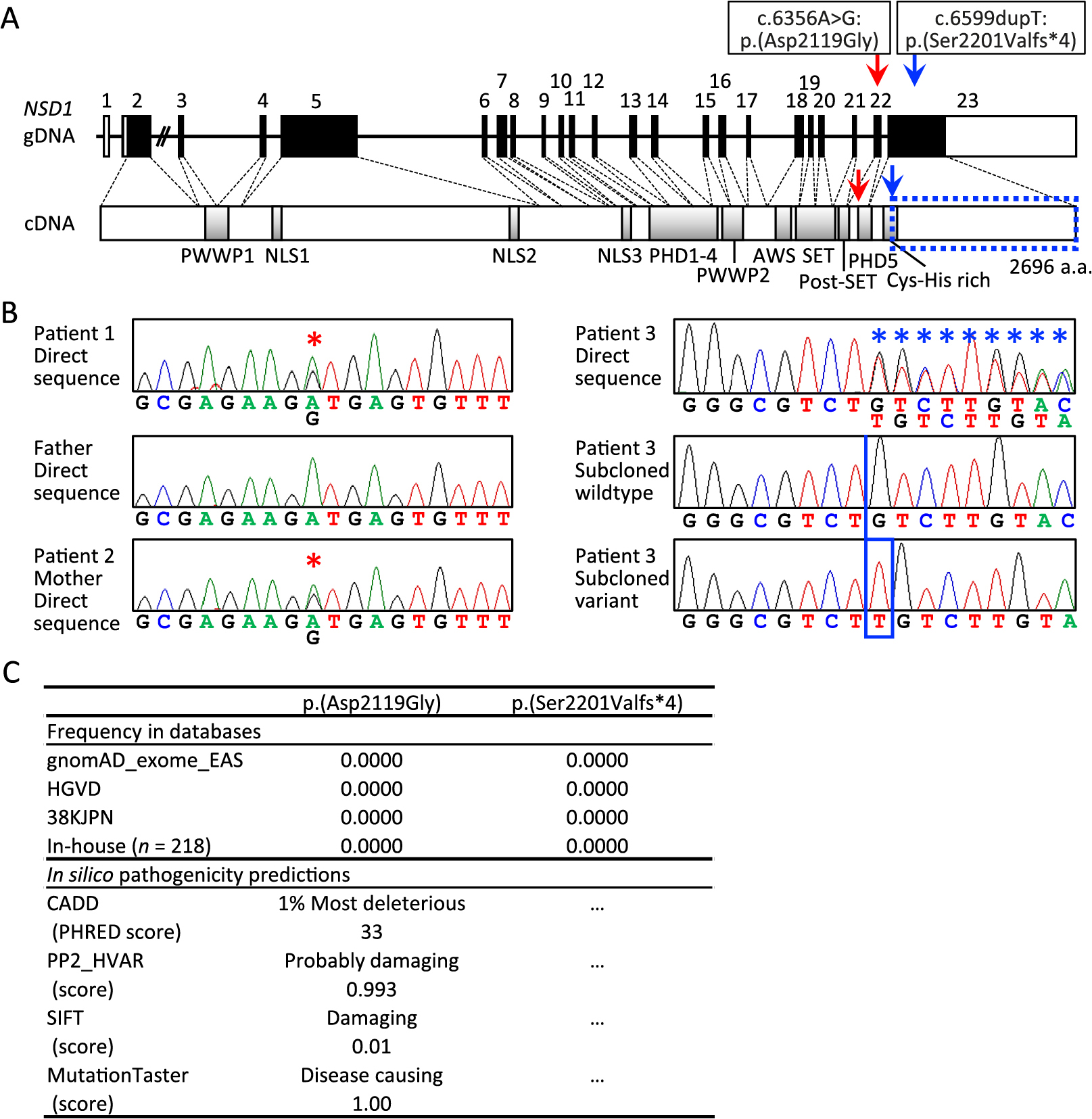
Consequently, we identified heterozygous rare NSD1 variants in patients 1–3, i.e., NM_022455.5:c.6356A>G:p.(Asp2119Gly) present in patients 1 and 2 and absent from the father of patient 1, and NM_022455.5:c.6599dupT:p.(Ser2201Valfs*4) present in patient 3 and absent from the mother of patient 3 (Fig. 2A). These variants were confirmed by Sanger direct sequencing and/or by sequencing of subcloned wildtype and variant alleles (Fig. 2B). Both variants were absent from all the public and in-house databases utilized in this study (Fig. 2C). The p.(Asp2119Gly) missense variant was predicted to have high pathogenicity (Fig. 2C), and the p.(Ser2201Valfs*4) frameshift variant on the last exon was predicted to escape nonsense mediated mRNA decay and to produce a truncated protein missing a part of Cys-His rich domain (Fig. 2A). No other rare variant was detected in growth-related genes of patients 1–3.
Discussion
Patients 1–3 showed Sotos syndrome-like overgrowth, macrocephaly, and characteristic appearance, but IDDs remained at a borderline level in patient 1 and was apparently absent in patients 2 and 3. According to the Japanese diagnostic criteria for Sotos syndrome (https://www.nanbyou.or.jp/entry/4778), the definitive diagnosis of Sotos syndrome can be established when a heterozygous disease-causing sequence variant in NSD1 or a microdeletion involving NSD1 has been identified in subjects with three major clinical features, i.e., macrocephaly (≥+2 SD) in infancy and toddlerhood, overgrowth (≥+2 SD) in infancy and toddlerhood, and characteristic appearance (e.g., large and long head, large hands and feet, protruding forehead and mandible, high arched palate, downward slanting palpebral fissures, and hypertelorism) (furthermore, if no causative genetic aberration has been identified, the clinical diagnosis of Sotos syndrome can be made in subjects with the above three major features plus IDDs). Thus, to confirm the diagnosis of Sotos syndrome, we performed genetic analysis, thereby revealing rare heterozygous NSD1 variants in patients 1–3. According to the ACMG guideline [6], the NSD1 variant identified in patients 1 and 2 is assessed as “likely pathogenic,” because it is positive for PS1 (same amino acid change as a previously established pathogenic variant), PM2 (absent from controls), and PP3 (multiple lines of computational evidence in support of a deleterious effect). Similarly, the NSD1 variant found in patient 3 is also assessed as “likely pathogenic,” because it is positive for PVS1 (frameshift variant predicted to escape NMD and to remove >10% of protein) and PM2. Collectively, the above clinical and genetic findings demonstrate the diagnosis of Sotos syndrome in patients 1–3.
Patients 1–3 had marked overgrowth with the SDS ranging from +4.1 to +6.3. In this regard, we performed a thorough review of the literature and found such extreme tall stature (≥+4.0 SD) in nine patients with molecularly confirmed Sotos syndrome (Table 2) [7-10]. Here, several findings are notable. First, while patient 1 was still in the early childhood, patient 2 had extreme tall adult height, and patient 3 showed overt overgrowth even as an adult. Similarly, while six of the previously reported nine patients with overt overgrowth were in their childhood, one patient aged 17 years and two adult patients exhibited extreme tall stature. This indicates the presence of Sotos syndrome patients with extreme tall stature in adulthood, although overgrowth is known to become infrequent and inconspicuous in adulthood [1, 3]. Second, the 12 patients had sequence variants rather than microdeletions affecting NSD1. This would be co-incidental, because the frequency of overgrowth is similar between patients with sequence variants and those with microdeletions [11]. In addition, since the variants reside at various domains in the 12 patients, it is unlikely that the sequence variants have some specific effects on growth. Third, serum IGF-1 was quite low in patient 1 and at the low-normal range in patient 3, consistent with the previous data indicating low serum IGF-I and IGF-II levels in patients with heterozygous NSD1 pathogenic variants [12]. This may be explained as a compensatory physiological reaction to decelerate the excessive growth caused by NSD1 aberrations, as discussed previously [12].
Table 2
NSD1 variants identified in patients with Sotos syndrome and excessive tall stature
|
Age |
Sex |
Height (SD) |
NSD1 variant* |
Domain |
| Pt 1 in this paper |
2 y 8 m |
M |
+4.4 |
c.6356A>G |
PHD5 |
| Pt 2 in this paper |
Adult |
F |
+4.1 |
c.6356A>G |
PHD5 |
| Pt 3 in this paper |
12 y 10 m |
F |
+6.3 |
c.6599dupT |
Cys-His rich |
| Pt 3 in ref. [5] |
1 y |
F |
+4.1 |
c.3141delC |
... |
| Pt 11 in ref. [5] |
9 y 6 m |
F |
+6.3 |
c.5386G>T |
PWWP2 |
| Pt 12 in ref. [5] |
0 y 3 m |
F |
+5.2 |
c.5398insT |
PWWP2 |
| Pt 15 in ref. [5] |
6 y 6 m |
F |
+4.1 |
c.6013C>T |
SET |
| Pt 19 in ref. [5] |
4 y |
F |
+5.0 |
c.3541delGAAA |
... |
| Pt 1 in ref. [6] |
17 y |
M |
+4.2 |
c.6523T>A |
Cys-His rich |
| Pt 1 in ref. [7] |
1 y 4 m |
M |
+4.0 |
c.896delC |
... |
| COG1879 in ref. [8] |
46 y |
F |
+6.0 |
c.6483G>A |
PHD5 |
| COG0107 in ref. [8] |
23 y |
F |
+4.0 |
c.6559C>T |
Cys-His rich |
Abbreviation: Pt, patient; y, year; m, month; M, male; F, female.
* According to NM_022455.5.
Several findings other than overgrowth are also worth pointing out in this study. First, IDDs remained at a borderline level in patient 1 and were apparently absent from patients 2 and 3, although they exhibited Sotos syndrome-compatible overgrowth, macrocephaly, and characteristic features. This would not be surprising, because it has been reported that 16–18% of adult patients with Sotos syndrome are free from IDDs [10, 13], and that IDDs tend to be milder in patients with NSD1 sequence variants than in those with microdeletions involving NSD1 [14]. Second, cardiac and renal anomalies as well as scoliosis were absent from patients 1–3. This would not be unpredicted because such features remain rather infrequent in patients with Sotos syndrome, especially in those with NSD1 sequence variants [13, 15]. Lastly, patient 2 was fertile in the presence of NSD1 likely pathogenic variant. This implies that NSD1 variants permit fertility at least in several patients, although a low reproductive rate has been suggested in patients with Sotos syndrome [10]. In this context, Laccetta et al. have suggested that NSD1 variants from exon 20 to exon 23 may not affect reproductive fitness [16]. Thus, the position of the variant in patient 2 may be relevant to the preserved fertility.
Conclusion
We identified NSD1 variants in three patients with extreme overgrowth. This implies that NSD1 should be examined in patients with marked overgrowth as the cardinal feature.
Acknowledgments
We thank Ms. Aya Kitamoto, and Ms. Fumiko Kato for their technical support.
Disclosure
The authors declare no conflict of interest associated with this research.
Funding Information
This work was supported by the grant from Japan Agency for Medical Research and Development (AMED) (JP23ek0109549).
References
- 1 Tatton-Brown K, Cole TRP, Rahman N (2004) Sotos Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, et al. (eds) GeneReviews. University of Washington, Seattle, USA: 1993–2023.
- 2 Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, et al. (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 30: 365–366.
- 3 Agwu JC, Shaw NJ, Kirk J, Chapman S, Ravine D, et al. (1999) Growth in Sotos syndrome. Arch Dis Child 80: 339–342.
- 4 Manor J, Lalani SR (2020) Overgrowth syndromes-evaluation, diagnosis, and management. Front Pediatr 8: 574857.
- 5 Masunaga Y, Nishimura G, Takahashi K, Hishiyama T, Imamura M, et al. (2022) Clinical and molecular findings in three Japanese patients with N-acetylneuraminic acid synthetase-congenital disorder of glycosylation (NANS-CDG). Sci Rep 12: 17079.
- 6 Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424.
- 7 Türkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, et al. (2003) Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 11: 858–865.
- 8 Zechner U, Kohlschmidt N, Kempf O, Gebauer K, Haug K, et al. (2009) Familial Sotos syndrome caused by a novel missense mutation, C2175S, in NSD1 and associated with normal intelligence, insulin dependent diabetes, bronchial asthma, and lipedema. Eur J Med Genet 52: 306–310.
- 9 Höglund P, Kurotaki N, Kytölä S, Miyake N, Somer M, et al. (2003) Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet 40: 51–54.
- 10 Foster A, Zachariou A, Loveday C, Ashraf T, Blair E, et al. (2019) The phenotype of Sotos syndrome in adulthood: a review of 44 individuals. Am J Med Genet C Semin Med Genet 181: 502–508.
- 11 Nagai T, Matsumoto N, Kurotaki N, Harada N, Niikawa N, et al. (2003) Sotos syndrome and haploinsufficiency of NSD1: clinical features of intragenic mutations and submicroscopic deletions. J Med Genet 40: 285–289.
- 12 De Boer L, Van Duyvenvoorde HA, Willemstein-Van Hove EC, Hoogerbrugge CM, Van Doorn J, et al. (2004) Mutations in the NSD1 gene in patients with Sotos syndrome associate with endocrine and paracrine alterations in the IGF system. Eur J Endocrinol 151: 333–341.
- 13 Saugier-Veber P, Bonnet C, Afenjar A, Drouin-Garraud V, Coubes C, et al. (2007) Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum Mutat 28: 1098–1107.
- 14 Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, et al. (2005) Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet 77: 193–204.
- 15 Machida M, Katoh H, Machida M, Miyake A, Taira K, et al. (2021) The association of scoliosis and NSD1 gene deletion in Sotos syndrome patients. Spine 2021 46: E726–E733.
- 16 Laccetta G, Moscuzza F, Michelucci A, Guzzetta A, Lunardi S, et al. (2017) A novel missense mutation of the NSD1 gene associated with overgrowth in three generations of an Italian family: case report, differential diagnosis, and review of mutations of NSD1 gene in familial Sotos syndrome. Front Pediatr 5: 236.

