REVIEW
Exploring mechanisms of insulin action and strategies to treat diabetes

2024 Volume 71 Issue 7 Pages 651-660

Details
2024 Volume 71 Issue 7 Pages 651-660
Insulin is a hormone that positively regulates anabolism and cell growth, whereas diabetes mellitus is a disease characterized by hyperglycemia associated with impaired insulin action. My colleagues and I have elucidated multifaceted insulin action in various tissues mainly by means of model mice. In the liver, insulin regulates endoplasmic reticulum (ER) stress response during feeding, whereas ER stress ‘response failure’ contributes to the development of steatohepatitis comorbid with diabetes. Not only the liver but also the proximal tubules of the kidney are important in the regulation of gluconeogenesis, and we revealed that insulin suppresses gluconeogenesis in accordance with absorbed glucose in the latter tissue. In skeletal muscle, another important insulin-targeted tissue, impaired insulin/IGF-1 signaling leads not only to sarcopenia, an aging-related disease of skeletal muscle, but also to osteopenia and shorter longevity. Aging is regulated by adipokines as well, and it should be considered that aging could be accelerated by ‘imbalanced adipokines’ in patients with a genetic background of progeria. Moreover, we reported the effects of intensive multifactorial intervention on diabetic vascular complications and mortality in patients with type 2 diabetes in a large-scale clinical trial, the J-DOIT3, and the results of subsequent sub-analyses of renal events and fracture events. Various approaches of research enable us of endocrinologists to elucidate the physiology of hormone signaling, the mechanisms underlying the development of endocrine diseases, and the appropriate treatment measures. These approaches also raise fundamental questions, but addressing them in an appropriate manner will surely contribute to the further development of endocrinology.
Insulin is a hormone secreted from pancreatic β cells that positively regulates anabolism and cell growth, and insulin receptor and the downstream transducer molecules have been identified in recent decades [1-4]. Impaired insulin action, however, leads to diabetes mellitus, a disease that is characterized by hyperglycemia and is associated with various complications and comorbidities that can impair quality of life and longevity. Now it is considered important not only to prevent diabetic complications but also to pay close attention to diabetic comorbidities which are more frequent in the elderly [5]. Here I review recent advances in the elucidation of the multifaceted roles of insulin, which contribute to the maintenance of nutritional homeostasis and consequently protection against diabetic comorbidities. I also review recent evidence on the prevention of vascular complications and mortality by multifactorial intervention in patients with type 2 diabetes.
Dynamic changes in nutrient metabolism are induced in the liver during the transition between fasting and feeding, and insulin is one of the major regulators with insulin receptor substrates (IRS)-1/2 as the key signaling transducer proteins. However, dysregulation of such processes may lead to diabetes and non-alcoholic fatty liver disease (NAFLD) [3, 6, 7], with metabolic dysfunction-associated steatotic liver disease (MASLD) as a newly proposed term [8].
My colleagues and I recently reported that, in the liver during feeding, insulin not only promotes protein synthesis but also regulates responses to endoplasmic reticulum (ER) stress, which is induced in parallel with an increased load of protein synthesis, based on findings in various mouse models [9]. Some of the responses during feeding are regulated by a transcription factor, X-box-binding protein-1 (XBP1)-s, which is encoded by spliced form of Xbp1 (sXbp1) mRNA and whose nuclear translocation is promoted by insulin signaling [10, 11]. Moreover, stromal cell-derived factor 2 like 1 (Sdf2l1), first reported as a protein up-regulated by ER stress [12], is regulated by XBP-1s and is deemed to be the key molecule in terminating feeding-induced ER stress and consequently maintaining metabolic homeostasis. We also revealed that Sdf2l1 interacts with transmembrane emp24-like trafficking protein 10 (TMED10), a protein involved in insulin secretion in pancreatic β cells [13], and promotes ER stress-associated degradation (ERAD) [9], by partially mimicking the roles of the orthologues in yeast [14, 15]. However, in cases of obesity and diabetes, insufficient insulin action leads to decreased levels of nuclear XBP-1s, down-regulation of downstream Sdf2l1, and consequently ER stress ‘response failure’, which further exaggerates ER stress and consequently insulin resistance in the liver. Such a vicious cycle is successfully treated by restoration of downstream Sdf2l1 expression, but not by over-expression of upstream XBP-1s, suggesting the possibility of Sdf2l1 as a potential therapeutic target for diabetes and comorbid NAFLD [9].
Moreover, analysis of human liver biopsy samples revealed that, in patients with diabetes, insulin resistance leads to insufficient induction of Sdf2l1 mediated by XBP-1s, which was suggested by a lower SDF2L1/sXBP1 mRNA ratio [9]. The insufficient induction is in turn correlated with staging of non-alcoholic steatohepatitis (NASH), with metabolic dysfunction-associated steatohepatitis (MASH) as a newly proposed term [8], or hepatic fibrosis. These associations were not observed in those without diabetes, probably because insulin resistance is canceled by, for example, compensatory insulin secretion, suggesting that NASH in patients without diabetes could be different from NASH in those with diabetes [9]. When we consider the classical two-hit hypothesis of NASH [16] in patients with diabetes, our results also imply that not only ER stress but also ER stress ‘response failure’ are deemed to be among the second hits to contribute to the development of NASH. Moreover, dysfunction of XBP-1s is expected to serve as a link between a major first hit, insulin resistance [17], which is decompensated due to comorbid diabetes, and an emerging second hit, ER stress ‘response failure’, and the SDF2L1/sXBP1 mRNA ratio is now one of the promising biomarkers of NASH comorbid with diabetes [9, 18]. Fig. 1 summarizes how impaired insulin action in the liver contributes to hyperglycemia and the progression of steatohepatitis in patients with type 2 diabetes.

Schematic description of potential pathophysiologic roles of impaired insulin action in the liver. Insulin resistance in the liver is not compensated in patients with type 2 diabetes, resulting in impaired insulin action, which in turn contributes to hyperglycemia and the progression of steatohepatitis.
In a nationwide survey in Japan, the fibrosis-4 (FIB-4) index, a hepatic fibrosis marker widely used in clinical practice, was reported to serve as a prognostic factor for the development of hepatocellular carcinoma (HCC) comorbid with type 2 diabetes [19]. Moreover, it was recently reported that gut insulin action is protective against the development of HCC in model mice [20], further suggesting the benefits of maintaining insulin action in patients with diabetes [18, 21]. It should be also noted that more attention is now focused on the concept of ER stress ‘response failure’ [18, 22], the roles of Sdf2l1 in a model rat of diabetes [23] and Crohn’s disease in humans [24], as was discussed previously [25], and those of TMEDs in various human diseases [26].
Gluconeogenesis in the liver plays a pivotal role in supplying glucose to glucose-utilizing tissues in a fasted state, but gluconeogenesis in the proximal tubules of the kidney also matters, especially during prolonged fasting [27, 28]. Insulin receptor is abundantly expressed in the cortex of the kidney, mainly consisting of the proximal tubules [29], and indeed, deletion of insulin receptor in the proximal tubules causes hyperglycemia [30]. My colleagues and I recently reported that gluconeogenesis in the proximal tubules of the kidney is subject to dual regulation not only by insulin but also by glucose [31].
We focused on IRS-1/2 [2, 3], and generated the proximal tubule-specific Irs1/2 knockout mice, a model of insulin resistance in the tissue generated by crossing Irs1/2-floxed mice [6] and sodium-glucose cotransporter 2 (SGLT2)-Cre transgenic mice that we newly generated independently [31] by following a previous report [32]. It was shown that gluconeogenesis is suppressed by insulin signaling mediated by IRS-1/2 in the proximal tubules, and impaired insulin signaling in the tissue leads to systemic insulin resistance [31]. We also showed that inactivation of the forkhead box O 1 (FoxO1), one of the key downstream molecules of insulin signaling [3, 33], via interaction with peroxisome proliferator-activated receptor gamma coactivator (PGC) 1α, is important in this context [31], although the contribution of the mammalian target of rapamycin (mTOR) pathway was also proposed recently [28]. Insulin receptor is considered to be expressed in the basolateral side of the proximal tubules [34], but glucose reabsorbed by SGLT1/2 expressed on the luminal side of the proximal tubules was also shown to suppress gluconeogenesis via suppression of Sirtuin 1 activity (Fig. 2). It seems that the proximal tubules sense primary urine glucose levels but do not care for blood glucose levels. Interestingly, gluconeogenesis is suppressed by SGLT inhibition even in models of impaired insulin action, suggesting that insulin signaling (intrinsic hormone signaling) could be overwhelmed by glucose signaling (rather extrinsic nutrient signaling), contrary to our assumption, at least in some tissues and in some conditions [31, 35].

Schematic description of molecules involved in the regulation of gluconeogenesis and renoprotective roles of adaptive response in the proximal tubules of the kidney. AMPK, 5' adenosine monophosphate-activated protein kinase; FoxO1, forkhead box O 1; IRS, insulin receptor substrate; PGC, peroxisome proliferator-activated receptor gamma coactivator; SGLT, sodium-glucose cotransporter.
Induction of gluconeogenesis is considered to be a kind of response that allows cells to cope with external stressors and survive. Such adaptive response is mainly mediated not only by Sirtuin 1 activated in response to under-nutrition but also by 5' adenosine monophosphate-activated protein kinase (AMPK) activated in response to hypoxia in the proximal tubules of the kidney. We and others propose that induction of adaptive response could be important for SGLT2 inhibitors to exert their renoprotective effects [36, 37], especially against hypoxia-induced tubular injury [38, 39], as was reviewed previously [25]. Moreover, the pro-gluconeogenic aspects of SGLT2 inhibitors might contribute to prevention of hypoglycemia [31], which could also be clinically important (Fig. 2).
Skeletal muscle is another insulin-targeted tissue of importance, especially in an absorptive state [40], and an aging-related disease in skeletal muscle is known as sarcopenia, a word derived from ancient Greek [41-44]. It is the established theory in the field of aging that sarcopenia is promoted by suppression of insulin/insulin-like growth factor (IGF)-1 signaling at least in lower organisms, such as yeast, nematodes, and flies [45], but in model mice and humans, impaired insulin action has been rather reported to be associated with muscle loss [46-49]. Furthermore, replacement of IGF-1 itself was one of the promising therapeutic approaches to sarcopenia [42].
My colleagues and I thus hypothesized that sarcopenia could be accelerated by suppressed insulin action in skeletal muscle associated with aging. We focused on Akt, the key downstream kinase of insulin/IGF-1 signaling [2, 3], and generated the skeletal muscle-specific Akt1/2 double knockout (mAktDKO) mice, as a model of aging-induced insulin resistance in the tissue [50]. As they age, the mice show reduced fast-twitch muscle mass, reduced body weight, impaired glucose uptake mediated by insulin and consequent systemic insulin resistance, weaker grip strength, and shorter exercise endurance, proving that these mice serve as a good model of sarcopenia. They also show suppressed osteogenesis and consequent osteopenia [50], an aging-related phenomenon in the bone that is known to co-exist frequently with sarcopenia in humans [51, 52]. Moreover, their lifespan is significantly shorter by around 10% with an increase in deaths from debilitation. Thus, insulin resistance in skeletal muscle leads to aging not only of skeletal muscle itself but also of the whole body [50].
Caloric restriction is generally believed to prolong longevity even in mammals [45], but the mAktDKO mice are found to be an exception in that they show accelerated reduction in body weight and die within just weeks of caloric restriction. Moreover, the mAktDKO mice fed with a high-fat diet as a model of sarcopenic obesity show still reduced lifespan with tumor death as the main cause. Consistently, subcutaneously transplanted melanoma cells grow faster in the mAktDKO mice. These results give us insights of clinical importance about diet therapy in patients with diabetes comorbid with sarcopenia [50].
Among these phenotypes observed in the male mAktDKO mice, those in skeletal muscle are replicated in the female mAktDKO mice but those in the bone or those that are systemic are not, suggesting the existence of sex differences in the impact of dysfunction of skeletal muscle on other tissues [50].
To explore key molecules downstream of Akt, we focused on the mTOR pathway, which is inactivated by deletion of Akt1/2 via enhanced activity of tuberous sclerosis complex 2 (TSC2) [3], with potential importance in skeletal muscle as was recently proposed [53], and the FoxO pathway, which is activated by deletion of Akt1/2. By comparing the phenotypes of the mAktDKO mice with those of the skeletal muscle-specific Akt1/Akt2/Tsc2 triple knockout (mAkt/TscTKO) mice and those of the skeletal muscle-specific Akt1/Akt2/Foxo1/Foxo4 quadruple knockout (mAkt/FoxoQKO) mice, the FoxO pathway is deemed to play pivotal roles in the development of osteosarcopenia and systemic aging due to inactivation of Akt, although the mTOR pathway is involved mainly in the mitochondria quantity control in skeletal muscle [50].
Curiously, the impact of impaired insulin/IGF-1 signaling in mammalian skeletal muscle on aging is diametrically opposite to that in the established theory in lower organisms [45], although they share the key downstream molecules, Akt and FoxO (Fig. 3) [50, 54]. Now the FoxO pathway is considered one of the promising targets in treatment of type 2 diabetes [55], but it should be also noted that the mTOR pathway was reported to be the key pathway downstream of Akt in macrophages and in the gut, at least in some conditions [20, 56].
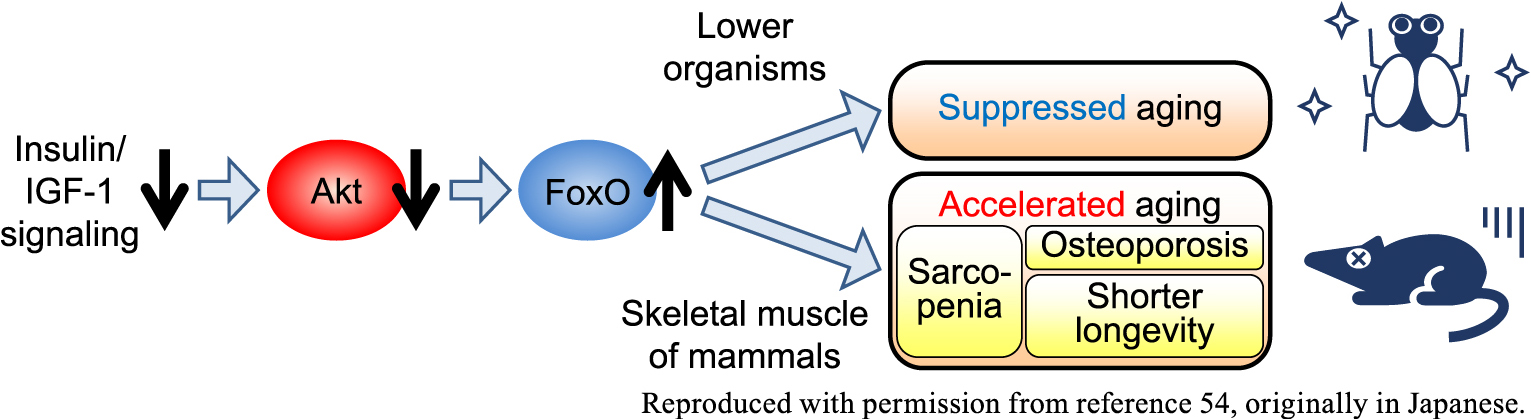
Schematic description of the opposite effects of insulin/IGF-1 signaling on aging in lower organisms vs. skeletal muscle of mammals, adapted with permission from reference 54, originally in Japanese. IGF-1, insulin-like growth factor-1.
Given that other skeletal muscle-specific knockout mice were recently reported as showing osteosarcopenia [57, 58], it can be fairly said that skeletal muscle serves as a kind of armor, just as those of gladiators in ancient Rome, against the development of frailty and aging [59], at least in males, as was reviewed recently [25].
Among the hormones to regulate aging and longevity other than insulin/IGF-1 is adiponectin [60]. My colleagues and I recently experienced a case with acquired generalized lipodystrophy treated with leptin replacement therapy for more than fifteen years who presented with severe aortic stenosis, which required transcatheter aortic valve implantation even in her thirties [61]. Given her genetic background [62], a variant in the LMNA gene (p.T10I) that causes generalized lipodystrophy-associated progeroid syndrome (GLPS) [63, 64], she might have been susceptible to progeria-associated disorders, such as aortic stenosis, an aging-related disease frequently observed in the elderly [65]. It is deemed likely that such susceptibility was accelerated by the prolonged ‘imbalanced adipokines’ over years associated with a lower circulating level of anti-inflammatory adiponectin due to lipodystrophy and higher circulating level of proinflammatory leptin due to sustained leptin replacement therapy (Fig. 4), although the treatment did lead to a definite improvement in her prognosis [61, 66]. In order to maintain a balance between the adipokines, co-replacement of adiponectin [67] or co-administration of an adiponectin receptor-activating antibody [68] could be a better approach rather than leptin replacement alone. More importantly, this case raises questions for us of endocrinologists as to whether replacement of a single hormone is sufficient to complement the function of a single endocrine organ or not and how the deficiency of the endocrine organ should be treated.

Schematic description of the proposed mechanisms underlying the development of severe aortic stenosis during leptin replacement therapy for generalized lipodystrophy-associated progeroid syndrome due to an LMNA variant in our case, adapted with permission from reference 66, originally in Japanese.
We also experienced another case with acquired generalized lipodystrophy due to recurrent panniculitis [69]. During the several weeks after initiation of corticosteroid treatment for recurrence of panniculitis and comorbid anemia, she showed rapid reductions in body weight and lean mass by 15% at maximum. Although she was still in her twenties, she had multiple risk factors for hypercatabolism and sarcopenia [42], lack of metabolic reserves, insulin resistance [50] and hyperglycemia [70] due to lipodystrophy, lowered daily activity due to anemia, and persistent inflammation and wasting associated with panniculitis. Relatively insufficient energy and protein intake during hospitalization is also deemed to have accelerated onset and development of corticosteroid-induced myopathy. Moreover, the lessons from this case could be generalized to show that careful attention should be paid to the potential liability to hypercatabolism, especially in skeletal muscle, in patients at high risk, such as those with diabetes, decreased metabolic reserves, and, potentially, malignancy, who are treated with drugs which could promote catabolism, such as corticosteroid and, potentially, SGLT2 inhibitors [69]. A potential benefit of SGLT2 inhibitors for lipodystrophy was proposed previously [71, 72], but a benefit-risk balance should be considered especially in the long run.
We also experienced a case of diabetes associated with myotonic dystrophy type 1 [73], a case of hyperinsulinemia associated with a variant in the IR gene [62], and two cases of hemoglobinopathies (Hb Hoshida [74] and HbJ Cape Town), each of which gives us intriguing clinical insights [25].
Clinical trials are powerful tools in providing us with answers to important clinical questions of the era [75], and the recent ones in the field of diabetes include how to prevent vascular complications, especially macrovascular complications. Among those undertaken in Japan in which patients with type 2 diabetes were registered is the Japan Diabetes Optimal Integrated Treatment study for 3 major risk factors of cardiovascular diseases (J-DOIT3). It is a randomized trial originally launched as a ‘strategic research’ program sponsored by the Ministry of Health, Labour and Welfare, in which 2,540 patients with type 2 diabetes were followed up in 81 facilities all over Japan for a median of 8.5 years [76-79]. The primary outcome was occurrence of any of a composite of myocardial infarction, stroke, revascularization, and all-cause mortality. Compared to conventional therapy recommended mainly by the guideline for the treatment for diabetes in Japan [80], intensive multifactorial intervention was shown to be associated with a non-significant risk reduction in the primary outcome, which was significant after adjustment for prespecified baseline risk factors. Intensive therapy was also associated with significant risk reductions in cerebrovascular events in a post hoc breakdown of the primary outcome and renal events and eye events among the secondary outcomes (Fig. 5) [79, 81]. Hypoglycemia was reported more frequently in the intensive therapy group, but it is noteworthy that the incidence of severe hypoglycemia was quite low and less than 1% in both groups [79].
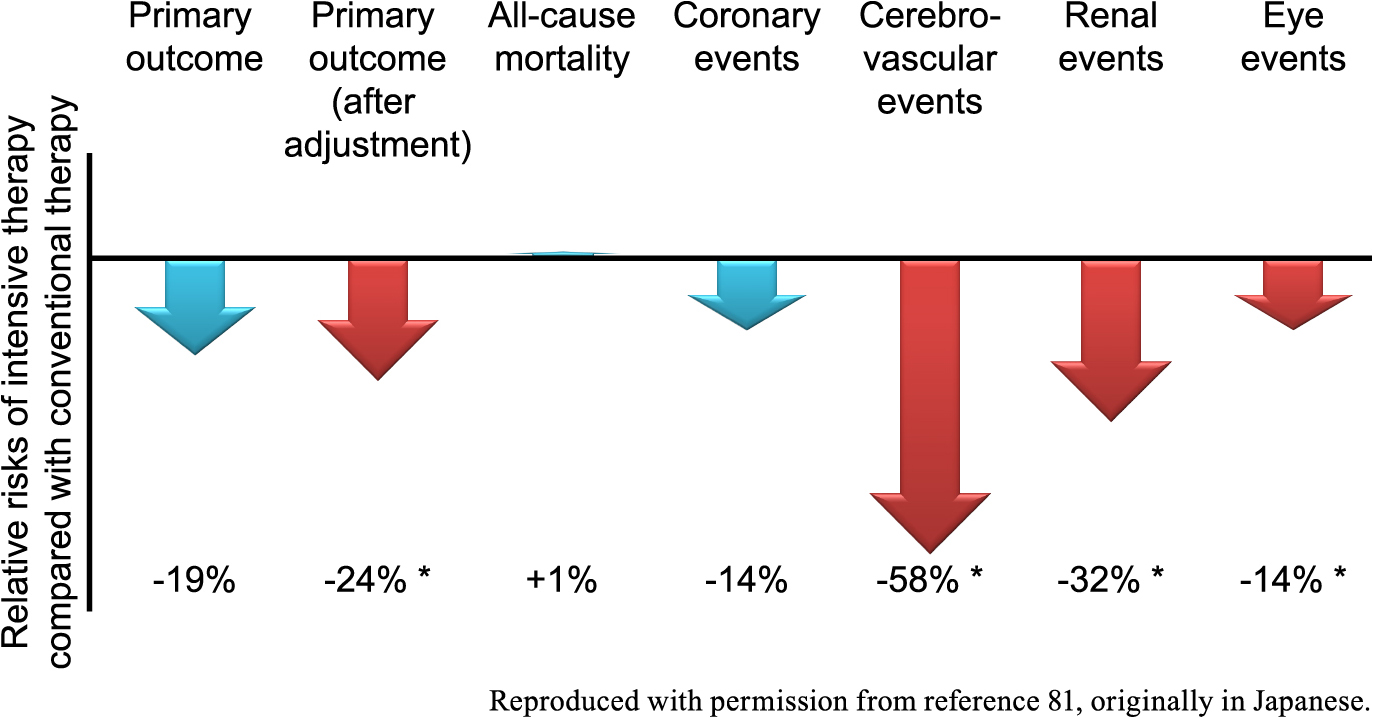
Summary of the effects of intensive therapy on the outcomes in the J-DOIT3, adapted with permission from reference 81, originally in Japanese. The primary outcome consists of all-cause mortality, coronary events, and cerebrovascular events, and the result was adjusted for baseline risk factors. Renal events and eye events are among the secondary outcomes. Effects of statistical significance (p < 0.05, shown with an asterisk) are shown in red arrows and others are shown in blue arrows. J-DOIT3, the Japan Diabetes Optimal Integrated Treatment study for 3 major risk factors of cardiovascular diseases.
The incidences of macrovascular complications in the J-DOIT3 turned out almost half those observed in the Japan Diabetes Complications Study (JDCS), which was started ten years earlier, despite similar baseline characteristics [82], suggesting the success of the current standard of diabetes care, at least by diabetes specialists and educators, in preventing macrovascular angiopathies [79]. Similarly, the incidences were shown to be much lower in the Anglo-Danish-Dutch Study of Intensive Treatment In People with Screen-Detected Diabetes in Primary Care (ADDITION-Europe) [83], which was launched in the early 2000s, compared to the United Kingdom Prospective Diabetes Study (UKPDS) [84], whose participants were recruited mainly in the 1980s, although these trials shared the key inclusion criteria of patients with newly diagnosed type 2 diabetes [85].
A subsequent sub-analysis of renal events revealed that glycemic control is associated with onset of nephropathy, or progression from normoalbuminuria to microalbuminuria, which comprised most of the renal events. It was also shown that blood pressure control is associated with estimated glomerular filtration rate (eGFR) decline in patients with a lower eGFR [79, 86].
It is also important to monitor adverse events carefully in clinical trials [87], and in terms of safety, my colleagues and I performed a sub-analysis of fracture, showing that intensive therapy was not associated with a significant risk elevation in fracture events [88]. Further, history of smoking in males and the FRAX (Fracture Risk Assessment Tool) score [89] and administration of pioglitazone in females were identified as risk factors for fracture comorbid with type 2 diabetes [88].
Among the three representative clinical trials to examine the effects of multifactorial intervention on diabetic complications in patients with type 2 diabetes, the mortality rate and the ratio of cardiovascular deaths to all deaths were found to be lower in the J-DOIT3 than in the other two trials [79, 90], the Steno-2 study [91] and the ADDITION-Europe [83]. From the viewpoint of causes of death and quality of life, diabetic comorbidities, such as malignancy, fracture, sarcopenia, and cognitive dysfunction, are deemed to be increasingly important [92]. The post-intervention observational study of the J-DOIT3 is under way [77], which will surely give us insights into the long-term multifaceted effects of intensive multifactorial intervention [25].
Various insights come from basic research, case studies, and clinical trials, enabling us to understand the physiology of hormone signaling, the mechanisms underlying the development of endocrine diseases, and the appropriate methods for treatment. Moreover, these approaches of research raise fundamental questions, such as whether hormone signaling could be always dominant over nutrition signaling and how the function of an endocrine tissue should be restored. These questions are expected to be addressed by us of endocrinologists, which will surely contribute to our better understanding of the endocrine system and improved care of endocrine diseases.
This review is essentially a summary of my presentation in the Japan Endocrine Society (JES) Research Award Lecture at the 96th Annual Congress of the JES, Aichi, Japan. I would like to express sincere gratitude to Dr. Takashi Kadowaki, Dr. Kazuyuki Tobe, Dr. Kohjiro Ueki, Dr. Naoto Kubota, and Dr. Toshimasa Yamauchi. I would like to express gratitude also to my colleagues and collaborators, the members of the Young Endocrinologist Conference (YEC) of the JES, and the members of the JES who have supported the YEC.
This article does not contain any studies with human or animal subjects performed by the author.
I declare that I have no conflicts of interest.