神経系疾患は,中枢神経(脳・脊髄),末梢神経,および筋肉を侵す疾患群であり,脱髄疾患,変性疾患,脳血管障害,脳外傷,脳腫瘍,先天性疾患,神経・筋疾患などがある。多くは難治・進行性であり,原因が不明で有効な治療法に欠ける。近年,家族性神経変性疾患のいくつかで原因遺伝子の同定が果たされるなど発症機序に関して遺伝子・蛋白質レベルの研究が相当進展してきており,病理組織学的診断の重要性が増している。髄鞘が破壊されるギランバレー症候群は末梢神経の代表的脱髄疾患である。神経変性疾患には,パーキンソン病,アルツハイマー病,脊髄小脳変性症,筋萎縮性側索硬化症などがある。脳腫瘍は頭蓋内(大脳,小脳,脳幹,硬膜,くも膜,血管,下垂体,および左右12対ある脳神経など)にできた腫瘍をいう。基本的に脳の組織から発生する原発性脳腫瘍と,他の臓器の腫瘍が脳に転移した転移性脳腫瘍に分ける。原発性脳腫瘍は,さらに良性の脳腫瘍と悪性の腫瘍に分類されるが,脳腫瘍には他のがんのようにTNM分類やステージ分類といったものはなく,悪性度(グレード)によって分類される。脳腫瘍の種類は星細胞腫,髄膜腫,神経鞘腫,下垂体線種は良性に分類されるが,星細胞腫と髄膜腫は悪性に変わることがある。毛様細胞性星細胞腫,乏突起膠細胞系腫瘍,膠芽腫や髄芽腫は悪性に分類される。膠芽腫と髄芽腫はグレードIVに分類されている脳腫瘍である。転移性腫瘍では肺癌,乳癌,前立腺癌,リンパ腫などが多い。
Nervous system diseases are a group of diseases that affect the central nervous system (brain and spinal cord), peripheral nerves, and muscles, including demyelinating diseases, degenerative diseases, cerebrovascular disorders, cerebral injuries, brain tumors, congenital neurological diseases, and nerve and muscle diseases. Many of these diseases are intractable and progressive, with their causes unknown and no effective treatments available. Recently, studies on the pathogenesis at gene and protein levels have progressed considerably. For example, the genes responsible for some familial neurodegenerative diseases have been identified. Accordingly, the importance of histopathological diagnosis has been growing. Guillain-Barre syndrome, which is characterized by the degradation of the myelin sheath, is a typical demyelinating disease of peripheral nerves. Neurodegenerative diseases include Parkinson’s disease, Alzheimer’s disease, spinocerebellar degeneration, and amyotrophic lateral sclerosis. Brain tumors develop in the skull (cerebrum, cerebellum, brainstem, dura mater, arachnoid membrane, blood vessels, pituitary gland, and 12 pairs of cranial nerves connected to the left and right hemispheres). Brain tumors are classified into primary brain tumors, which basically originate in the brain tissue, and metastatic brain tumors, which originate in other organs and spread to the brain. Primary brain tumors are further classified into benign and malignant brain tumors. Different from other cancers, there is no tumor, nodes, and metastases (TNM) classification or staging system for brain tumors. Brain tumors are classified according to their grade of malignancy. Astrocytoma, meningioma, schwannoma, and pituitary adenoma are classified as benign brain tumors. However, astrocytoma and meningioma may undergo malignant transformation. Pilocytic astrocytoma, oligodendroglial tumor, glioblastoma, and medulloblastoma are classified as malignant brain tumors. Glioblastoma and medulloblastoma are classified as grade IV brain tumors. Metastatic brain tumors commonly originate from lung cancer, breast cancer, prostate cancer, and lymphoma.
病原体として細菌,真菌,ウイルス,原虫,寄生虫等が挙げられ,感染経路としては中耳炎や副鼻腔炎などの隣接病巣からの直接波及や血行性,さらに手術後を含む外傷性などがある。真菌や寄生虫感染は免疫不全状態にある患者でみられることが多い1),2)。
2) 画像および肉眼所見一般的に周囲に浮腫を伴った多発性の病変を形成する。膿瘍を伴う感染巣ではMRIでリング状の増強効果を伴う占拠性病変を形成するため悪性脳腫瘍との鑑別が問題となる。肉眼的に軟らかい病変であることが多いが,肉芽腫性変化が強いと弾性硬を示す。
3) 基本的な組織像病原体がウイルスである場合,炎症細胞の主体はリンパ球で,血管周囲での集簇が観察される。一方,細菌あるいは真菌の場合には好中球,組織球を主体とした炎症細胞浸潤が著しく,壊死を伴った膿瘍を形成する。特に結核菌や真菌感染においては壊死周囲に毛細血管の増生を伴った肉芽腫形成がみられ,多核巨細胞が混在する(Figure 13.1)。また感染巣内およびその周囲では泡沫組織球の集簇や反応性星細胞増生(グリオーシス)が目立つ(Figure 13.2)。グリオーシスとは感染,梗塞,脱髄や種々の腫瘍辺縁部でみられる反応性の変化で,星細胞系腫瘍との鑑別が問題となることが多い。腫瘍性星細胞との鑑別で最も重要な所見は細胞密度とその分布である。腫瘍では毛細血管や神経細胞周囲に密に集簇する傾向があるが,グリオーシスでの細胞密度は疎で,均等に分布し,豊富な細胞質と線維性突起が明瞭である1),2)。

血管周囲性リンパ球浸潤(左)と壊死を伴う肉芽組織(右)。

泡沫組織球の浸潤・集簇(左)と反応性星細胞の増生(右)。

リンパ球と共に多数の泡沫組織球を認める(左)。放射状に伸長する細胞質突起が明瞭な星芒状の反応性星細胞(右)。
多くの泡沫組織球とリンパ球や好中球などの炎症細胞浸潤を認め,背景にはフィブリン析出を伴った壊死物質を認める。ウイルス感染では神経細胞や膠細胞の核にすりガラス状のクロマチン変性や核内封入体構造を認めることがある。また四方八方に伸長する細長い線維性突起が目立つ反応性星細胞の増生を認める。グリオーシスにおける星細胞は腫瘍性星細胞と比較して星芒状の細胞質がより広く,線維性突起が豊富で,多核細胞は稀である2),3)。
2. アスペルギルス感染(aspergillus infection) 1) 概要アスペルギルス種は環境中で最もよくみられる糸状菌の一つで原因菌はAspergillus fumigatusが多い。免疫能が低下した患者で,気管支拡張症や肺気腫などに起因する肺の空洞や副鼻腔または外耳道など開放腔に感染する傾向がある。HIV感染などの免疫不全状態では侵襲性かつ破壊性で,全身の血行性播種を生じることがある。アスペルギルスの中枢神経への感染経路は約8割が肺などの遠隔病巣からの血行性播種,残りが副鼻腔などの近接臓器からの連続的侵襲あるいは手術などを含む外傷による直接感染といわれている。頭蓋底浸潤のアスペルギルス症は重症化する症例が多い。血清中のβ-D-グルカンの上昇も重要な参考所見となる1),2)。
2) 画像所見感染が進行すると肉芽腫あるいは空洞を伴った膿瘍を形成する。その場合,造影MRIで強いリング状の増強効果を示すため,悪性脳腫瘍や脱髄性疾患との鑑別が必要となる(Figure 13.4左)。

左:造影MRIでリング状に増強される嚢胞様病変。
右:壊死巣と周囲の泡沫組織球を含む炎症細胞浸潤。
膿瘍,壊死を伴う肉芽腫を形成することが多い(Figure 13.4右)。炎症細胞の主体は好中球であることが多いが,多くのリンパ球および泡沫組織球も混在する。アスペルギルスの菌糸は45度のY字状に分岐する特徴があり,隔壁を有し,幅は3~6 μm幅で,樹枝状に伸長する1)~3)。ヘマトキシリン(時にエオジン)に淡染し,PAS反応ならびにGrocott染色は菌糸の同定に有用である(Figure 13.5)。

Y字状に分岐し,隔壁を有する菌糸はヘマトキシリンに淡染し,PAS反応に陽性を示す。
圧挫標本でも好中球,泡沫組織球を含む壊死性の背景に分枝する菌糸が観察される(Figure 13.6)。アスペルギルスの菌糸は,カンジダ属の菌糸よりも太く,ムコール属よりも細く,幅がより均一な特徴があるが,実際の病原真菌の同定には細菌学的検査が不可欠である3)。

好中球と泡沫組織球を背景にY字分岐する菌糸を認める。
クリプトコッカスはHIV感染者や強力な免疫抑制治療を受けている患者(臓器移植後やサルコイドーシスなど)で発症することが多い。原因菌としてはCryptococcus neoformansが最も多く,鳩の糞で汚染された土壌中に豊富に存在し,菌の吸入により肺に感染する。通常,吸引された菌体は肺胞マクロファージにより貪食・排除されるが,一部は休眠酵母状態として潜伏的に感染する日和見感染の形態をとる。免疫抑制状態になると菌は再活性化し,肺炎などの症状が引き起こされる。中枢神経系に移行しやすい傾向があるため,進行すると髄膜炎や脳脊髄炎が併発する。クリプトコッカス脳脊髄炎は永続的な神経障害を引き起こすことがあり,致死率は10%程度といわれている1)~3)。
2) 画像所見菌体は血管周囲腔(Virchow-Robin腔)や脳脊髄液内で増殖し,くも膜下腔に沿って進展する。結果的に血管周囲腔が拡張し,偽嚢胞を形成する。偽嚢胞はMRI-T1強調で低信号,T2強調で高信号を示す。感染が進行すると脳血管関門が破壊され脳実質内に進展し,稀であるが肉芽腫性の病変が形成される。この場合には,リング状の増強効果を示すことがあるため,悪性脳腫瘍との鑑別が問題となる。
3) 組織像菌体は5~20 μmの円形の酵母様真菌で,肉芽組織中の多核組織球の細胞質内に貪食された形で観察されることが多い(Figure 13.7)。ムチカルミン染色で莢膜は赤色に陽性を示すが,菌体は染色されない1)~3)。
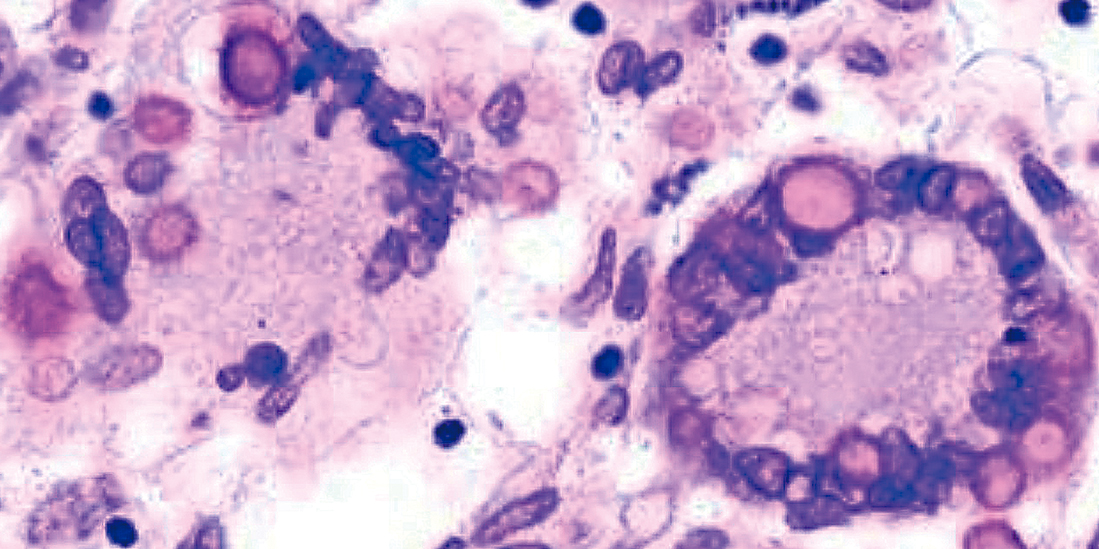
多核組織球の細胞質内に大小の酵母様真菌を認める。
組織標本と同様,菌体は多核組織球の細胞質内で観察されやすい。HE染色ではエオジンに,Pap染色ではライト緑に淡染する二重のリング状を示す莢膜を有する(Figure 13.8)。髄液中のクリプトコッカスの証明には墨汁染色が有用である。菌体および莢膜には墨汁粒子が入り込めないため色素が抜ける像を示す(Figure 13.9)。しかし,墨汁染色では炎症細胞もクリプトコッカスに類似した像を示すため誤認しないように注意する必要がある。

HEでエオジンに,Papでライト緑に淡染する二重のリング状を示す莢膜を有する円形の酵母様真菌。

クリプトコッカス菌体および莢膜には墨汁粒子が入り込めないため色素が抜ける(神戸常盤大学 畠榮先生より提供)。
トキソプラズマ症(toxoplasmosis)は人畜共通感染症で,Toxoplasma gondiiの感染によって発症する。通常は無症候性感染であるが免疫不全状態で再活性化し,様々な重篤な症状が引き起こされる。AIDS患者における多発性脳病変の最も多い原因菌で,HIV感染発見の契機となることも稀ではない。発症様式としては急性から亜急性,慢性の経過を辿るものまで様々で,症状としても発熱,頭痛,痙攣,認知障害など多彩である1)~3)。血清学的検査でのトキソプラズマIgG,IgM抗体価の上昇は重要な参考所見となるが,PCR法による髄液中のT. gondii DNA検出が最も特異度が高く,確定診断のカギとなる。
2) 画像所見MRIではT2/FLAIRで境界不明瞭な高信号を示し,造影MRIでリング状に造強される多発性腫瘤を形成する(Figure 13.10)。病変は灰白質と白質の境界部,大脳基底核,脳幹,小脳に多く,画像診断では,しばしば悪性リンパ腫や転移性脳腫瘍との鑑別が問題となる1),2)。

T2/FLAIRで境界不明瞭な高信号(左),ガドリニウムでリング状の増強を伴う不整形の腫瘤性病変(右)が多発。
著明な壊死を伴う膿瘍形成とその周囲にグリオーシス,好中球浸潤,ミクログリアの増生がみられる。トキソプラズマ菌体は壊死周囲のviableな組織に認めることが多く,小型のコンマ状を示す急増虫体(tachyzoites)ならびに無数の緩増虫体(bradyzoites)を含み,薄い膜で囲まれた50~70 μm大の球状のシストがみられる。これら虫体の確認が診断に不可欠で,免疫染色による同定が可能である(Figure 13.11)1)~3)。

無数の緩増虫体を含む球形のシストを認める(左)。
免疫染色にてシスト,嚢子が陽性を示す(右)。
フィブリンの析出ならびに著しい好中球浸潤を伴った汚い壊死性の背景に,泡沫組織球ならびに反応性星細胞の増生を認める。急増虫体および緩増虫体を含むシストが確認できれば確定診断が可能である3)。画像診断上,悪性リンパ腫を含む原発性脳腫瘍が鑑別として問題となるが,明らかな異型細胞を欠くため,鑑別に苦慮することは少ない。むしろ,その他の感染症との鑑別が問題となることが多いため,免疫化学染色を含めた特殊染色の併用に加え,種々の病原体に対する血清中の抗体価などの臨床情報を十分に把握することが重要である。
5. 進行性多巣性白質脳症(progressive multifocal leukoencephalopathy; PML) 1) 概要Progressive multifocal leukoencephalopathy(PML)は,潜伏感染しているJCウイルス(JCV)が重篤な免疫不全状態で再活性化し,髄鞘を形成する乏突起膠細胞に感染することで脳内に脱髄巣が多発する疾患である。免疫不全をきたす基礎疾患としてはHIV感染,造血器系悪性腫瘍や膠原病などが代表的である。典型的には大脳の皮質と白質の境界部~皮質下白質に小さい脱髄巣が多発し,これらが互いに融合しながら拡大していく。臨床的には片麻痺,四肢麻痺,認知機能障害や失語などが初期症状であるが,進行が極めて速く,発症後速やかに免疫機能の回復がなされないと予後不良で,多くの場合が1年以内に死亡するといわれている1),2)。
2) JCウイルスポリオーマウイルス属の二重銀環状DNAウイルスで,PML患者より分離されたのが最初で,患者氏名のイニシャルからJCウイルスと命名された。ポリオーマウイルス属には他にSV40ウイルスやBKウイルスがある。JCVの初感染は無症候であるが,腎臓の集合管上皮細胞や骨髄および末梢血中のB細胞に潜伏持続感染していると考えられている2)。
3) 画像所見MRIではT2強調およびFLAIR像で高信号を示す大小の病変が皮質下白質に多発する。通常,浮腫は認めず,増強効果に乏しいが,時にリング状の増強効果を伴うこともある(Figure 13.12左)。
4) 組織像白質内に島状の脱髄斑が散在する脱髄性疾患に類似した組織像を呈する(Figure 13.12右)。泡沫組織球ならびに反応性星細胞の増生もみられるため他の感染性脳炎や低悪性度の星細胞腫との鑑別が問題となる(Figure 13.13)。血管周囲性のリンパ球浸潤がみられることもあるが,免疫不全状態を反映して他のウイルス感染症と比較して明らかにその頻度は低い1),2)。PML診断に重要な所見はJCVに感染した乏突起膠細胞の同定で,これらの細胞は核が明らかに腫大し,顆粒状あるいは泡沫状の核内封入体構造を有し,時にすりガラス状のクロマチンを呈する(Figure 13.14)。JCVの存在はin situ hybridization法あるいはPCR法によって確定されるが免疫化学染色も有効で,感染細胞はJCV後期蛋白であるAgnoprotienおよび粒子外殻(カプシド)蛋白であるVP1に陽性を示す。JCV早期蛋白であるLTに反応するSV40-T antigenならびにp53蛋白にも陽性を示すが,宿主蛋白との交差反応を示すこともあるためその特異度は低い(Figure 13.15)1),2)。
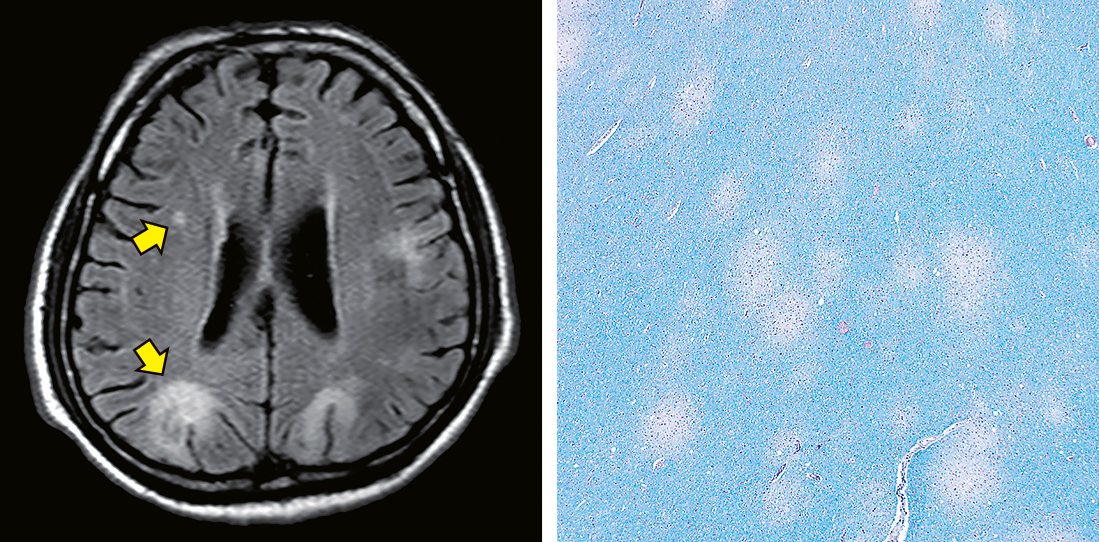
T2/FLAIRで高信号を呈する白質に多発する病変(左)。
組織では島状に多発・散在する脱髄斑を認める(右)。

多数の泡沫組織球と反応性星細胞の増生を認める。

多数の泡沫組織球,反応性星細胞を背景に核が膨化し,顆粒状の封入物を有する乏突起膠細胞(矢印)を認める。

感染細胞はAgnoprotien(左)およびVP1に陽性を示す(中)。またLTに反応するSV40-T antigenにも陽性を示す(右)。
圧挫標本においてはその他の感染症と同様,多数の泡沫組織球の浸潤ならびに集簇とグリオーシスが基本的な細胞像である(Figure 13.16)。組織標本と同じくJCV感染細胞の同定が重要で,核が明らかに腫大し,ドット状の封入体構造を含むすりガラス状クロマチンパターンを呈する乏突起膠細胞の認識がPML診断の指標となる(Figure 13.17)。核異型を伴った星細胞が目立つ場合,星細胞腫との鑑別が問題となるが,グリオーシスにおける星細胞では長い線維性の細胞質突起を放射状に伸長する傾向があり,N/Cも細胞密度も低い。また多数の泡沫組織球が腫瘍性病変で出現することは稀である。臨床的にPMLが疑われる症例では,強拡大下での細胞の核所見を詳細に観察することが重要である1)~3)。

泡沫組織球とリンパ球を伴ったグリオーシスを背景に核が膨化し,すりガラス状を示す乏突起膠細胞(矢印)を認める。

腫大した核内にドット状の封入体を含み,すりガラス状を呈する乏突起膠細胞の出現がPML細胞診断の指標となる。
腫瘤形成性脱髄病変(tumefactive demyelinating lesion; TDL)は,脱髄病変が散発する多発性硬化症(multiple sclerosis; MS)の中でも2 cm以上の限局性腫瘤を形成し,臨床症状としても麻痺,失語,認知障害や限局性神経脱落症候など脳腫瘍に類似した症状を呈する疾患で,画像診断においても原発性脳腫瘍との鑑別が問題となる3)~5)。20~30代の若年者に好発し,女性にやや多い。基本的にはステロイドパルス療法が行われるため,治療方針が全く異なる脳腫瘍や感染症との鑑別診断は極めて重要である。
2) 画像所見前頭・頭頂葉を中心としたテント上にみられ易い。周囲に広範な浮腫を伴う結節状の病変で,灰白質に向かって開放する不完全なリング状の増強効果を伴うことが多い(open-ring sign)。MRS(MR spectroscopy)は腫瘍と脱髄病変の鑑別に有効で,脱髄巣では炎症に起因する細胞膜の破壊に関係するコリン(Cho)の上昇と,ニューロン障害を反映するN-acetyl aspartate(NAA)の低下によりCho/NAAが上昇する。しかし,この所見は神経膠腫の中でも壊死を伴う膠芽腫ではみられるため注意が必要である(Figure 13.18)。

広範な浮腫を示す高信号域を認め,内部には低信号の小斑状の病変を伴う(左)。MRSではCho/NAAが正常と比較して上昇する(右)。
脱髄病変における基本的な組織所見は,①白質に散在する大小の脱髄斑(Figure 13.19),②血管周囲性のリンパ球浸潤,③グリオーシス(Figure 13.20),④泡沫組織球の集簇(Figure 13.21)である。これらの所見はPMLに代表されるウイルス感染症に酷似しており,免疫不全をきたす基礎疾患の有無などの臨床情報を把握することに加え,神経細胞や神経膠細胞の核内における封入体構造やクロマチン変性の有無を詳細に観察することが重要である。脱髄巣内では多少の膨化変性をきたした軸索を認めるものの,神経原線維は保持されているため,neurofilament proteinおよびBodian染色に陽性を示す線維構造がみられる(Figure 13.22)。この点が梗塞巣とは異なる4),5)。感染症と同様,反応性星細胞に様々な核異型を伴うため星細胞系腫瘍との鑑別が問題となるが,泡沫組織球と小リンパ球からなる多彩な炎症細胞浸潤および集簇巣の形成は,腫瘍では一般的にみられない所見である。

淡明化した大小の島状に点在する脱髄斑がみられる。

血管周囲に集簇するリンパ球浸潤像が目立つ。背景には種々の程度で異型を示す反応性星細胞の増生を認める。

崩壊した髄鞘成分である脂質を貪食した泡沫組織球ならびにミクログリアの集簇巣を認める。

脱髄巣内では少数の膨化変性した軸索を認めるものの,基本的に軸索は保たれている。この点が脳梗塞とは異なる。
細胞診標本においても,豊富な神経膠線維性背景(glio-fibrillary background)を形成する反応性星細胞の増生と毛細血管周囲に集簇あるいは散在性に多数の小型リンパ球浸潤を認める(Figure 13.23)。また数多くの泡沫組織球がみられ,これらがシート状集塊を形成して出現することもある。反応性星細胞も種々の程度で核異型を示し,TDLに特異的ではないが一部に多数の小型核を有する多核巨細胞(Creutzfeldt細胞)や顆粒状を示す核分裂像(starburst mitosis)がみられることがある(Figure 13.24)。

神経膠線維性の背景に反応性星細胞の増生を認め,血管周囲に集簇および散在性に多数の小リンパ球浸潤を認める。

多数の泡沫組織球とリンパ球浸潤(左)。豊富な細胞質突起を有する反応性星細胞が増生し,ときに多数の小型核を有する大型の多核星細胞(Creutzfeldt細胞:右図矢印)を認める。
悪性リンパ腫では血管周囲性にリンパ球の増殖がみられるため鑑別診断の対象となる。強拡大での個々の細胞の核形不整や核小体の有無などの詳細な観察が重要である。中枢神経系原発の悪性リンパ腫は大細胞型B細胞性リンパ腫が圧倒的に多く,核異型が強く,明らかに細胞サイズが大きい。PMLをはじめとする種々のウイルス感染との鑑別は神経細胞および神経膠細胞の核所見(封入体構造,すりガラス状クロマチン)が重要なポイントである。星細胞腫との鑑別に関しては,感染症の項で述べたように反応性星細胞は星細胞腫と比較して,四方八方に伸長する細長い線維性突起が目立ち,星芒状の細胞質は保持され,N/Cが低い。また非常に多くの泡沫組織球の出現は,より非腫瘍性病変を疑う所見である3)。その他に原発性中枢神経系血管炎や脱髄性疾患の一つである急性散在性脳脊髄炎(acute disseminated encephalomyelitis; ADEM)も鑑別疾患として挙げられるが,細胞診のみでの鑑別は不可能で,病歴,経過,画像所見などの臨床情報の把握が重要である4),5)。
近年,認知症の患者数は増加傾向にあり,なかでもアルツハイマー病(Alzheimer disease; AD)は最も多い原因疾患である。最初の症例は1906年にドイツの精神医学者Alois Alzheimerにより報告され,1910年にEmil Kraepelinにより「アルツハイマー病」と命名された。
大脳皮質の神経細胞を侵す変性疾患であり,神経細胞の脱落,老人斑と神経原線維変化などこの疾患を特徴づける組織所見がみられる。それらの主成分はアミロイドβとリン酸化タウである。初老期以降に記憶障害を主体とする症状があらわれて判断力や自発性の低下などを伴いながら経過とともに進行し,日常生活において常時介護が必要になる6)。ほとんどが散発性であるが同一家系内に複数のAD発症者を認める家族性ADから原因遺伝子としてPSEN1 (14q24.3), PSEN2 (1q31-q42), APP (21q21), APOE (19q13.2)の変異が同定された。また,ADに特徴的なバイオマーカーを取り入れて無症候期から病態の進展に対応した新しい診断基準が2011年にNational Institute of Aging/Alzheimer’s Associationのワーキングループによって提案され,発症前を3段階にわけている7)。認知症状を発症する10数年前より脳でのアミロイドβの蓄積が示されており,一期は無症候性アミロイド沈着,二期はアミロイドβ陽性でシナプス機能不全あるいは早期の神経細胞変性がおこり髄液中のタウ値が上昇,三期はアミロイドβ陽性で神経細胞変性があり軽微な認知機能低下のみられる時期に分かれる。バイオマーカーは大きく2種類に分かれる。一つはアミロイドβの沈着を示すバイオマーカー検査でありアミロイドPETによる脳内集積の陽性,髄液中ではアミロイドβ42の低値を示す。もう一方は,神経障害を示すバイオマーカー検査で,髄液中のタウならびにリン酸化タウの高値,MRIにより海馬の萎縮,18F-FDG-PETまたは脳血流SPECTにより側頭葉頭頂葉の機能低下などを知る方法である8)。
2) 肉眼所見典型例では脳皮質の著明な萎縮がみられ前頭葉,側頭葉,頭頂葉で著明であるが(Figure 13.25),肉眼的に萎縮が認められなくても顕微鏡像ではADに特徴的な組織所見が認められることがある。白質の容量も減少し脳室の拡張がみられるが,被殻や尾状核は比較的よく保たれている。

脳実質がびまん性に脱落することにより脳皮質は狭くなり脳回が狭小化して脳溝が拡大している。
大脳皮質にみられる神経原線維変化と老人斑はこの疾患に特徴的な組織所見である(Figure 13.26, 13.27)。正常高齢者の海馬,海馬傍回にもみられるがADでは基底核,視床,視床下核,マイネルト核,脳幹の一部でもみられ出現数と脳全体に占める割合が重要である。

神経細胞の細胞質内には神経原線維変化を生じて束ねられたように変性した線維が凝集しており好塩基性に染まっている(矢印)。背景には神経細胞は消失し,線維だけが変性して好酸性に染まり神経細胞があった跡としてみられる(矢頭)。

毛細血管の直上のニューロピルには,エオジンで濃く均一に染まる芯とそのまわりを放射状に伸びる構造からなる。星状膠細胞がそれを取り囲むように浸潤している。
HE染色では神経細胞内に好塩基性に染まり線維がもつれたneurofibrillary tangleとしてみられる。また,神経細胞の輪郭を残すように変性して好酸性を呈する神経原線維となり,崩壊した神経細胞の痕跡としてみられる。正常の神経原線維は微小管microtubuleと神経細線維neurofilamentからなるが,リン酸化タウの蓄積により正常の神経細胞ではみられないpaired helical filament(PHF)呼ばれる80 nmごとにくびれを持つ細管の束で構成されている9)。本来のタウは微小管重合促進因子として働き,3R(3 repeat)タウと4Rタウが存在しADではそれらがほぼ1対1で神経細胞に蓄積する(Figure 13.28)。神経原線維変化は嗅内野から海馬などの辺縁系を経て大脳新皮質へと進展していく10)。

神経細胞の細胞質には糸くず様にタウの沈着した神経原線維がからみ合って突起まで伸びている。神経細胞の間にみられる糸状のニューロピルスレッドも3Rタウと4Rタウの両方で茶色の陽性像が確認できる。
ニューロピルにはエオジンで一様に染まる芯を持ちその辺縁へ放射状に伸びるアミロイド線維からなる斑状の構造物としてみられる。斑の中心部分は密に凝集したアミロイドβからなる芯を有しており,周囲の明るい部分を隔ててアミロイド線維と神経原線維変化を伴った神経突起が蛇行しながら混在し沈着する(Figure 13.29)。その周りには小膠細胞や星状膠細胞が反応して集まっている(Figure 13.30)。アミロイド仮説ではAPP, PSEN1, PSEN遺伝子の変異によりアミロイド前駆タンパク質の産生と細胞膜内で切断が亢進しアミロイドβの産生量が増加する。さらに,アミロイドβがオリゴマー化してシナプス毒性を発揮し,なんらかの過程を経てタウを介した神経毒性につながり神経細胞を死に至らすと考えられている11)。老人斑は前頭葉と側頭葉底部の新皮質から出現して海馬体,扁桃体,間脳,基底核へと広がり,高度に進行した症例ではさらに中脳,下脳幹から小脳皮質にもみられる。

アミロイドβの沈着した陽性部分は茶色を示し,アミロイド芯と呼ばれる中心部は強陽性に染まる。芯を持たない老人斑や陽性顆粒がニューロピルへ沈着しているのもみられる。

写真真ん中よりやや左側に星状膠細胞の取り囲まれた老人斑がみられる。内部にはリン酸化タウの蓄積した太い神経突起の断片をみることが出来る。写真右側の神経細胞にはリン酸化タウが蓄積して神経原線維変化を伴ってみられる。
ボディアン染色は神経原線維,神経突起,軸索を明瞭に染められ,リン酸化タウの蓄積した異常な神経原線維も正常の神経線維と一緒にみることが出来る(Figure 13.31)。AD症例の染色では切片の厚さは6 μm程度だと神経原線維の変化が観察しやすい。

神経原線維変化のある神経細胞内には黒色に染まるneurofibrillary tangleを明瞭に観察することが出来る。写真中央では老人斑に混在する太い変性した神経突起が褐色に染まってみられる。
老人斑は黒色にコントラストよく染まり,弱拡でも容易にみることが出来る。神経原線維変化のある細胞も褐色~黒色に染めることが出来るのでADの組織所見を観察するのに有用な染色法である(Figure 13.32)。

老人斑は黒色に染まり低倍からでもハッキリと確認することが出来る。神経原線維変化のある細胞もコントラストよく染色される。
5%メセナミン水溶液で調整したメセナミン銀液で反応する方法13),ピリジン銀を前処理として行ったあとにPAM染色用メセナミン銀液で反応する方法14)が考案されている。
⑤ ガリアス・ブラーク染色リン酸化タウの蓄積している異常な神経原線維や神経突起あるいは星状膠細胞が黒く染色される。神経突起内に蓄積したリン酸化タウが黒く糸状に染まるニューロピルスレッドがみられる13)。星状膠細胞の線維全体が染まるものは房状アストロサイト,先端のみが染まるものはアストロサイト斑と呼ばれる(Figure 13.33)。

神経細胞内のneurofibrillary tangle,ニューロピルスレッド,房状アストロサイトの細い線維まで明瞭にみることが出来る。老人斑内のリン酸化タウが蓄積して神経原線維変化を伴った神経突起も黒く染め出される。
レビー小体型認知症(dementia with Lewy bodies; DLB)は比較的新しい疾患概念であり,小坂らが提唱したレビー小体病,びまん性レビー小体病などを基礎として1995年にNewcastle upon Tyneで開催された国際ワークショップで「レビー小体型認知症」の名称が提唱された15)。DLBはAD,血管性認知症とともに三大認知症といわれており,変性疾患では2番目に多い原因疾患である。早期ではADと比べて記憶障害は軽度であるが認知機能の低下は目立つことが多い。幻視,視空間・構成障害,日中の眠気と傾眠,レム睡眠行動障害,寡動,筋強剛,嗅覚の低下などの症状が認められる6)。
中枢神経系と自律神経系に多数のレビー小体ならびにレビー神経突起の出現が特徴である。レビー小体を有する神経細胞では細胞死を引き起こし16),軸索では神経伝達物質などの輸送を傷害することが明らかとなった。
それらの構成蛋白質であるαシヌクレインは,もともと老人斑を構成する非アミロイドβ成分として単離された蛋白である。神経系に広く発現しており,シナプス前終末に局在してSNAREタンパク質複合体の形成に際して分子シャペロンとして機能していると考えられている。
頭部MRIではADと比べて内側側頭葉の萎縮は軽度であり,中脳背側部が萎縮していると報告されている。DLBに特徴的な検査はFDG-PETで後頭葉に糖代謝の低下,ドーパミントランスポーターの123I-β-CITや123I-FP-CITを用いたSPECTで線条体での集積の低下,交感神経の機能をみる123I-MIBG心筋シンチグラフィーでは心筋へのMIBCの取り込みの低下が報告されている8)。
2) 肉眼所見海馬領域を含む側頭葉内側部の萎縮がみられるがADと比べて軽度である。その他には前頭葉の萎縮に進行すると大脳全体がびまん性に萎縮を示す。中脳の黒質や橋の青斑核ではメラニン含有神経細胞が消失し減少するため黒褐色の色調が失われる(Figure 13.34)。

健常人と比べてDLB症例では黒質の色調が薄くなっているのが分かる。
神経細胞の細胞質内あるいは神経突起内にレビー小体と呼ばれ,HE染色では好酸性に染まる芯を持ちその周囲には明瞭なハローを有する構造物である。電顕でも光顕と同じく2層あるいは3層の同心円状の構造をしている(Figure 13.35)。芯の部分は電子密度の高い微細顆粒,細線維,リング状のものが密集しており,外側は10 μm程度の弱々しく太さの不均一な細線維の集まりが放射状に配列し間には有芯小胞がみられる9)。この異常な細線維の構成成分がαシヌクレインであることが同定され,蓄積したαシヌクレインはリン酸化をされており抗リン酸化αシヌクレイン抗体を用いた免疫組織染色でレビー小体と,レビー関連ニューライトを観察することが出来るようになる17)(Figure 13.35)。レビー小体ではαシヌクレインにユビキチン,ニューロフィラメントなどの蛋白が結合している(Figure 13.36)。また,pale bodyはレビー小体の前段階でありそれを産生する母地であると考えられており,レビー小体よりも大きな類円形をしたαシヌクレイン陽性の構造物が細胞質内にみられる18)。

メラニン色素含有神経細胞の細胞質にレビー小体がみられる。中心部は好酸性に濃く染まり,その周りは明るいハローを有した2層構造をなしている。レビー小体の外側はリン酸化αシヌクレインで強陽性を示すが芯部分は染まらない。ニューロピルには好酸性に染まる太い神経突起もみられる。レビー関連ニューライトと呼ばれる神経突起もリン酸化αシヌクレインで陽性を示す。

HE染色ではエオジンに濃く染まる大小2つのレビー小体が神経細胞の細胞質内にみられる。ボディアン染色ではピック小体のような嗜銀性はみられない。リン酸化αシヌクレイン陽性,ユビキチン陽性を示す。
αシヌクレインの蓄積は最初に迷走神経背側核と嗅索で起こり脳幹では上向性に進行し扁桃体から新皮質へと拡がっていく。CDLBガイドラインではレビー小体の分布と出現数により脳幹優位型,辺縁型,新皮質型に分類している19)。レビー小体は迷走神経背側核,青斑核,黒質,扁桃体,マイネルト基底核,線条体,側頭葉の全内側部から側頭葉外側皮質,島回,帯状回などに出現し,その後に交感神経節や消化管神経叢へも出現するようになる(Figure 13.37, 13.38)。

神経細胞の細胞質内には丸く染まりの淡いpale body様の類円形の構造がみられる。外套細胞に囲まれた内部にもエオジンで淡く染まる構造物がみられる。

神経細胞の細胞質内の類円型構造物と外套細胞に囲まれた類円形構造物はリン酸化αシヌクレイン陽性を示す。
前頭葉側頭葉変性症(frontotemporal lobar degeneration; FTLD)はピック病を原型として,臨床的に前頭側頭型認知症,意味性認知症,進行性非流暢性失語といった3つの変性を伴う認知症を包括した疾患概念である。前頭葉・側頭葉に限局した病変を有し,性格変化,行動障害,言語障害などを主張とする非アルツハイマー型の認知症である6)。
FTLDの病理所見は多彩であり病因も単一ではない。病理学的には異常凝集して封入体を形成し,蓄積した蛋白質による分類が用いられている。
FTLDはAD, DLBに次いで3番目に多い認知症をきたす変性疾患であり,好発年齢は45~65歳,65歳以下ではADについで多い認知症といわれている。
2) ピック病 ① 肉眼的所見葉状萎縮と呼ばれる前頭葉と側頭葉に限局した高度の萎縮が特徴的であり20),皮質と白質の容量がともに減少し脳回は極端に薄くなりナイフエッジ様と表される(Figure 13.39)。

前頭葉と側頭葉の脳回が萎縮し脳溝が拡張してみられる。左右を半分ずつ隠して前後を見比べると分かりやすい。萎縮は皮質だけでなく白質の容積も減り,ナイフエッジ様と表されるほど脳回は極端に薄くなっている。
大脳皮質の神経細胞が脱落し,皮質と髄質に強いグリオーシスを生じる。残存した神経細胞質内にはピック小体と呼ばれる嗜銀性の円形をした封入体を有する(Figure 13.40)。ピック小体は大脳皮質の第2層,海馬歯状回の錐体細胞,線条体などの小型神経細胞に認められやすく,HE染色では淡い青色を呈して大きさは核と同じくらいである(Figure 13.41)。電顕では15 nmの真っ直ぐな線維と160 nm周期でねじれた線維からなり9),3Rタウと神経細線維を含んでいる。ピック小体を含み細胞質の片側が膨れて,ニッスル顆粒が不明瞭となった神経細胞はピック細胞と呼ばれている。嗜銀性のない封入体を含んだ神経細胞もピック細胞と呼ばれることがあるので区別する必要がある。

神経突起を有する神経細胞の細胞質内軸索起始部に好銀性を示す褐色のピック球がみられる。嗜銀性があることから嗜銀球とも呼ばれている。

神経細胞の細胞質内に淡く好塩基性に染まる封入体を含んで膨らんだピック細胞がみられる。
FTLDの大部分において神経細胞ならびに神経膠細胞の細胞質内に封入体が形成され,それを構成する蛋白質も明らかとなった。病的に意味のある蛋白質を基にFTLD-tau,FTLD-TDP(transactive response DNA binding protein 43 kDa, TDP-43),FTLD-FUS(fused in sarcoma),FTLD-UPS(ubiquitin proteasome system)と明らかな封入体を伴わないFTLD-ni(non inclusion)の5つに分類される21)。本項ではFTLD-tauとFTLD-TDPについて触れる。
① FTLD-tauタウの異常蓄積を伴うFTLDはFTLD中で約50%を占める。タウは微小管結合蛋白で微小管の重合促進と軸索では微小管の安定化に働き,微小管結合に関わる繰り返し配列の数により3Rタウと4Rタウというアイソフォームが存在する。タウ陽性封入体の発現様式により3Rタウのみが蓄積しているピック病,4Rタウのみの大脳皮質基底核変性症,進行性核上麻痺,嗜銀顆粒性認知症,グリア細胞球状封入体タウオパチー,3R/4Rタウの両方の神経原線維変化型痴呆などの3群に分類される。
病変部では広範なグリオーシスがみられ,ホルツァー染色では神経膠線維が密になり濃く染まっている部位と神経膠細胞が豊富な明るい部位とがみられる(Figure 13.42)。神経細胞の細胞質では4Rタウの蓄積がみられ免疫組織化学あるいはガリアス・ブラーク染色で封入体を確認することが出来る(Figure 13.43)。リン酸化タウが乏突起膠細胞の核の周囲を取り巻く様に蓄積してみられるものはグリアルコイル状小体,神経突起に蓄積してみられるものは嗜銀性スレッドと呼ばれている(Figure 13.44)。タウが星状膠細胞の突起の近位側に蓄積したものは房状アストロサイトと呼ばれ進行性核上性麻痺,遠位側に蓄積したものはアストロサイト斑と呼ばれ大脳皮質基底核変性症で出現する。

星状膠細胞の細胞質は染まらずに核と突起が青紫色に染まる。神経膠線維が密になり濃青紫色を呈する領域(矢印)とその脇には神経膠線維は比較的粗で星状膠細胞の豊富な領域がみられる。

神経細胞の細胞質内封入体は4Rタウ陽性,ガリアス・ブラーク染色で細胞質内に蓄積するタウが黒色に染まる。

4Rタウ陽性,ガリアス・ブラーク染色で黒色に染まるグリアルコイル状小体(矢印)がみられる。
TDP-43の異常蓄積を伴うFTLDは,FTLD中で約40%を占める。TDP-43は,核内リボ蛋白でRNAを標的としてスプラッシング,翻訳,輸送や安定化の制御に関わっている。正常細胞では核内に存在しているが,病的状態では細胞質内にリン酸化された状態で封入体を形成する。
FTLD-TDPと筋萎縮性側索硬化症(amyotrophic lateral sclerosis; ALS),その中間に位置するFTLD-MND(motor neuron disease)とは病理所見や原因遺伝子の共通性も明らかとなり一連のスペクトラムと広く捉えられている。
ALSの脊髄では前角の神経細胞が脱落し,髄鞘染色を行うと後索は比較的保たれているが側索と前索は太さの不均一な髄鞘からなり脱髄のように抜け落ちるのではなく明るくみえる(Figure 13.45)。前角では大型の神経細胞は脱落し,残存する神経細胞は萎縮して濃染あるいは虎斑融解を生じるものがみられる。変性した神経細胞の細胞質中には好酸性で類円形をした大きさ数μmのブニナ小体を含むものもある(Figure 13.46)。

後索は髄鞘が青く染まっているがそれ以外は広範に変性により太さは不均一になり髄鞘の崩壊しているところもあり染色性が弱くなっている。

大型の神経細胞は変性して脱落し,残存する神経細胞の細胞質には大きさは数μmで1個から数連なり好酸性で類円型をしたブニナ小体がみられる。
本疾患では残存する神経細胞の細胞質内に顆粒状あるいはスケイン状などの封入体22)がリン酸化TDP-43(pS409/410)で陽性を示す(Figure 13.47)。

神経細胞の細胞質内にリン酸化TDP-43陽性の辺縁不正形,顆粒状,スケイン様など様々な形の封入体がみられる。
びまん性星細胞腫は,星細胞に類似した形態を示す異型星細胞が脳実質にびまん性かつ浸潤性に増殖する腫瘍で,中でも形態学的に細胞異型のみがみられる場合をびまん性星細胞腫(WHO grade II)と分類される。さらに遺伝学的にIDH1/2遺伝子(イソクエン酸脱水素酵素遺伝子)変異の有無により大別され,変異を認める腫瘍はIDH変異型(IDH mutant),変異がない腫瘍はIDH野生型(IDH wild)に分類される。遺伝子解析が行われず情報が不十分な場合にはnot otherwise specified(NOS)を付記する(Figure 13.48)23)~27)。


左:MRI-T1強調で低信号を示す腫瘤。増強効果は乏しい。
右:肉眼像。多房性嚢胞を伴う境界不明瞭な灰白色腫瘤。
腫瘤はMRI-T1強調で低信号,T2強調で高信号を示し,造影剤による増強効果に乏しい。成人大脳半球特に前頭葉と側頭葉に好発する。肉眼的には境界不明瞭な灰白色で軟らかい。嚢胞を伴うこともある24)~26)。
3) 組織像(Figure 13.50)
細胞密度が高く,背景の膠線維性の細胞質突起が豊富。
正常白質に比べ明らかに細胞密度が高く,異型星細胞がびまん性に増殖する。核は類円形~短紡錘形で,クロマチンの増量,核形不整を示す。様々な量の線維性の細胞質突起を伸長するため,網目状の膠線維性背景(glio-fibrillary background)が目立つ。腫瘍細胞が神経細胞や血管周囲,軟膜下などに密に集簇するSchererの2次構造(secondary structure)がみられ,グリオーシスとの鑑別点となり得る。核分裂像は認められず,微小血管増殖や壊死もみられない3),23)~29)。
4) 免疫学的および遺伝子学的所見(Figure 13.51)
腫瘍細胞はIDH1(R132H)に陽性,ATRXの核発現が消失。
ほぼ例外なくglial fibrillary acidic protein(GFAP)が細胞質と突起に強陽性を示し,S100蛋白にも陽性。IDH変異型の90~95%がIDH1遺伝子の132番目のコドンのアルギニンがヒスチジンへの点変異R132Hであり,残りがIDH2遺伝子変異である。IDH1(R132H)変異は感度・特異度共に優れた免疫抗体により検出することができる。またATRX(α-thalassaemia/mental retardation syndrome X-kinked)遺伝子の変異が高率にみられ,免疫染色にて核での発現が消失する。またTP53遺伝子変異はびまん性星細胞腫の約半数の症例で認められ,免疫染色にて10%以上の細胞で核の強陽性を認める場合,TP53変異とみなすことができる。これらの免疫学的所見はびまん性星細胞腫とグリオーシスとの鑑別診断に有効である23)~26)。
5) 圧挫細胞像(Figure 13.52)
毛細血管周囲に集簇する線維性突起と異型核を有する細胞。
線維性細胞質突起が豊富な異型星細胞が毛細血管周囲に集簇するように分布する。核クロマチンの増量,形状不整,大小不同性を認めるが,極めて異型に乏しいこともある。反応性星細胞との鑑別が最も重要であるが,星細胞腫では細胞密度が高く,細胞質があまり目立たず,N/Cが高く,細胞質突起が太く短い。泡沫組織球が集簇することは稀である3),30),31)。
6) 肥胖細胞性星細胞腫(gemistocytic astrocytoma)偏在する核と好酸性で顆粒状の豊富な細胞質を有する肥胖細胞に類似した腫瘍細胞が増殖するびまん性星細胞腫の一亜型で,腫瘍細胞の20%以上が肥胖細胞よりなる腫瘍と定義される(WHO grade II)。組織および圧挫標本ともに大型の異型星細胞が集簇性に増殖する。線維性突起は太くて短いかほとんどみられない。一般的な星細胞腫に比べて核異型が強く,クロマチンの凝集,多核,多形性が目立つ(Figure 13.53, 13.54)。退形成性星細胞腫への悪性転化を起こしやすいことが知られている23)~29)。

偏在核と好酸性で広い細胞質を有する腫瘍細胞。

好酸性でやや顆粒状を示す細胞質は厚く,突起は太くて短い。二核細胞が目立つ。
びまん性星細胞腫と膠芽腫の中間的な増殖能と予後を示す腫瘍(WHO grade III)。先述のびまん性星細胞腫と同様,IDH遺伝子変異の有無により,IDH変異型,IDH野生型,NOSに分類される。びまん性星細胞腫から進展するものあり,さらに膠芽腫へと進展する傾向が強く,診断後2年以内に膠芽腫になるといわれている。画像診断的には部分的に増強効果を示すことが多い。組織および細胞学的には退形成性変化が強く,明らかに細胞密度が高く,クロマチンの粗大化や形状不整,大小不同などの多形性が増す(Figure 13.55)。核分裂像の数が重要な所見で,小さい組織片で1個以上認められたら本腫瘍と診断される(Figure 13.56)。毛細血管は増生するが,血管内皮細胞の多層性の増殖はみられず,壊死巣も認めない。この点が膠芽腫との重要な鑑別点である。しかし実際の術中迅速診断に際しては,限られた材料での両者の鑑別は極めて困難なため,high grade glioma(高異型度膠腫)として報告されることが多い23)~29)。

肥胖細胞様の異型星細胞が多く出現。細胞の大小不同性も目立つ(圧挫標本)。

明らかに細胞密度が高く,核の腫大,核形不整,クロマチン増量と粗剛化などの多形性が目立つ。核分裂像(矢印)の確認が本腫瘍の診断に重要(左:組織標本,右:圧挫標本)。
退形成性および増殖活性が著しく高く,臨床的にも最も高い悪性度を示す膠腫(WHO grade IV)。90%以上がIDH遺伝子変異を伴わないIDH野生型(原発性)膠芽腫で,残りがIDH変異を有するびまん性星細胞腫あるいは退形成性星細胞腫から悪性転化したと考えられるIDH変異型(二次性)膠芽腫である。原発性膠芽腫は50歳以降の高齢者に好発し,極めて予後不良で5年生存率は10%未満である23)~27)。
2) 画像・肉眼所見大脳半球(特に前頭葉,側頭葉)に好発し,MRIではリング状に強く増強される(Figure 13.57)。腫瘍は軟らかく,出血,壊死を伴い,周囲脳への広範な浸潤性増殖を示す。片側大脳半球の腫瘍が脳梁を介して対側に浸潤し,蝶型割面像(butterfly appearance)を示すことがある。近年,手術時の腫瘍部同定を目的とした5-アミノレブリン酸(5-ALA)蛍光診断が用いられている。事前に内服した5-ALAは腫瘍内に取り込まれ,プロトポルフィリンに代謝・蓄積される。これを400 nm青色励起光下で観察すると腫瘍部は赤色蛍光を発するため正常部と腫瘍部が明瞭に区別することができる(Figure 13.58)。一般的に増殖活性が高い領域ほど蛍光強度が強いため,術中迅速診断時のサンプリングにおいてその威力を発揮する。

左:造影MRI(T1強調)リング状に増強される腫瘤性病変。
右:肉眼像。出血・壊死を伴い広範に浸潤する腫瘍。

400 nmの青色励起光下で腫瘍部は赤色蛍光を発する。
組織像は多彩で紡錘形,多角形,類円形など多形性に富む異型が強い細胞が充実性に増殖する。一部には結合性を有し上皮様に見えたり,小型円形細胞よりなり悪性リンパ腫との鑑別が問題となるような症例もある。核は著しいクロマチンの増量,明瞭な核小体,核形不整が目立ち,多核や巨細胞などもしばしば観察される。核分裂像が散見され,異常な核分裂像もみられる(Figure 13.59)。基本的には星細胞への分化を示すため豊富な細胞質突起を有し,特に血管周囲でより目立つglio-fibrillary backgroundを形成する。腫瘍内には大小の壊死巣がみられ,これらを取り囲むように腫瘍細胞の核が柵状に配列する柵状壊死(palisading necrosis)を認める。また毛細血管増生も著しく血管壁の内皮細胞の核が腫大し,多層性に増殖する微小血管増殖像(microvascular proliferation)がみられる。いずれの所見も膠芽腫の特徴で本腫瘍診断の重要な根拠となる(Figure 13.60)。

多形性が強く,多核の腫瘍細胞や豊富な細胞質を有する上皮様の異型細胞もみられる。核分裂像が多数認められる。

壊死巣を囲む柵状壊死(左)と微小血管増殖(右)が特徴的。
亜型として,奇怪な形状を示す多核の巨細胞が主体をなす巨細胞膠芽腫,紡錘形細胞が線維肉腫様に増殖する部分がモザイク状に混在する膠肉腫,EMAやcytokeratinに陽性の上皮様細胞やラブドイド細胞が充実性に増殖する類上皮性膠芽腫などがある23)~27)。
4) 圧挫細胞像組織像と同様,異型性の強い細胞が高い細胞密度で出現する。膠線維が豊富で網目状の背景が目立つ(Figure 13.61)。個々の異型細胞は悪性と判断するのが容易なほど著しい核異型を示し,核分裂像も散見される。また壊死巣を反映して核崩壊物を含むデブリスを認める(Figure 13.62)。圧挫標本においても微小血管増殖像は観察可能で,内皮細胞核の腫大,重層化,不規則な蛇行がみられる(Figure 13.63)3),30),31)。

明らかに細胞密度が高く,多核の巨細胞も目立つ。豊富な線維性細胞質突起の存在は星細胞性腫瘍を疑う指標となる。

大小不同,核形不整,クロマチン粗大化を伴った異型細胞。核分裂像も散見され,背景には壊死を想起させる好酸性の無構造物質および小型濃縮核もみられる。

血管周囲にまとわりつくように増殖する腫瘍細胞(左)と,内皮細胞が重層し,壁が肥厚した毛細血管を認める。
近年の悪性膠腫(退形成性星細胞腫および膠芽腫)の治療法として,ニトロソウレア系アルキル化剤(BCNU wafer)脳内留置療法が行われるようになった(Figure 13.64)。本薬剤は腫瘍の摘出腔に留置されることで徐々にBCNUが浸透し,殺腫瘍効果を発揮する。膠芽腫では一般的に放射線治療が行われるが,術後から放射線治療開始までの期間を補うことができるため良好な治療成績が得られている。本薬剤の適応疾患は悪性膠腫であるため,術中迅速診断における低異型度(WHO grade I~II)と高異型度(WHO grade III~IV)の鑑別診断が求められるようになっている。従って,術中病理所見と臨床および画像診断に矛盾がある場合,特に悪性膠腫が疑われているのに低異型度病変しか認められない場合は,採取部位が腫瘍の辺縁部で代表的な病変が得られていない可能性が高いため,積極的に追加組織の提出を求めるなど,術者との綿密なコミュニケーションが重要である。また圧挫標本のサンプリングも非常に重要である程度の大きさの腫瘍材料が提出された場合は,肉眼所見を詳細に観察し,出血や壊死が疑われる部位,正常部と比べて色調が異なる部位など複数箇所から標本を作製するべきである30)。

ニトロソウレア系アルキル化剤(カルムスチン)が含まれた円盤状の薬剤を腫瘍の摘出腔内面に留置する治療法。
先述のびまん性星細胞腫と共に2016年WHO分類改定でその疾患定義が大きく変化した腫瘍である。本腫瘍は,乏突起膠細胞によく似た均一な腫瘍細胞がびまん性に増殖・浸潤する腫瘍で,IDH1/2遺伝子変異と1番ならびに19番染色体(1p/19q)の共欠失を示す腫瘍と定義される。この両遺伝子異常が証明された腫瘍がIDH変異・1p/19q共欠失型乏突起膠腫(WHO grade II)であり,それ以外の異常もしくは遺伝子解析が行われない腫瘍は乏突起膠腫NOSと呼ばれる。後述の組織学的基準を満たす退形成性所見を伴った腫瘍は退形成性乏突起膠腫と定義され(WHO grade III),IDH1/2および1p/19qの遺伝子情報あるいはNOSが付記される23)~27)。
2) 画像・肉眼所見MRIでは比較的境界明瞭な腫瘤で,T1強調で低~等信号,T2強調で高信号を呈し,CTではしばしば石灰化像を認める。造影効果は概して乏しい。退形成性乏突起膠腫では出血,壊死,嚢胞変性などを伴うため,内部が不均一な腫瘍として描出され,多くの症例で造影効果がみられる。肉眼的には灰白色あるいは赤桃色の軟らかい腫瘍である(Figure 13.65)。

T1強調で低~等信号を示す腫瘤。石灰化を伴うことが多い(左)。肉眼的に桃色がかった白色調の軟らかい腫瘍(右)。
びまん性星細胞系腫瘍と同一の腫瘍起源細胞にIDH遺伝子変異が生じ,次いで1p/19q遺伝子の共欠失が加わることで腫瘍が発生すると考えられている。この1p/19q共欠失は,1番染色体と19番染色体がお互いのセントロメア付近で相互転座を起こし,生じた派生染色体が細胞から脱落することに起因する。この証明にはFISH法が有用である(Figure 13.66)。一方,星細胞系腫瘍で高頻度にみられるTP53変異は本腫瘍では認めず相互排他的と考えられている。すなわちIDH変異が生じた共通の腫瘍起源細胞において二次的に起こる遺伝子変異がTP53であれば星細胞系腫瘍,1p/19qであれば乏突起膠細胞系腫瘍が形成されるという腫瘍発生モデルが考えられている。

1p/19q転座による派生染色体の脱落を反映してFISH法にて両染色体共に緑2個,赤1個の欠失パターンを示す。
本腫瘍における1p/19q共欠失は診断マーカーに留まらず,予後および治療効果予測因子としても重要で,PCV療法(P: procarbazine,C: CCNU,V: vincristin)に対する反応が良好といわれている23)~29)。
4) 組織像ほぼ正円形の核と明るい細胞質を有する腫瘍細胞が均一に増殖する。核周囲には固定および脱水による人工産物である明庭を認め,目玉焼き像(fried egg appearance)あるいは蜂の巣構造(honeycomb structure)と呼ばれる。背景には鶏小屋の金網像(chicken wire pattern)と表現される樹枝状の毛細血管網が発達し,微小石灰化を認めることが多い(Figure 13.67)。偏在核とGFAP陽性の好酸性細胞質を有するminigemstocytesも観察される(Figure 13.68)。大脳皮質への浸潤部では神経細胞の周囲に腫瘍細胞が取り囲む像perineuronal satellitosisがしばしば認められる。退形成性乏突起膠腫ではこれらの所見に加えて,細胞密度の増加,核小体の明瞭化などの核異型が増加する。診断基準としては「高倍率10視野中に6個以上の核分裂像」あるいは「微小血管増殖」のいずれかを認めるとされる。壊死に関しては乏突起膠腫でも稀に認めることが知られているが,退形成性乏突起膠腫の方が明らかに多く,柵状壊死を伴うことがある23)~27)。腫瘍細胞は高率にOlig2,Nkx2.2に陽性,星細胞系腫瘍で陰性化するATRXの核発現は保たれている(Figure 13.69)。

正円形核と明るい細胞質を有する腫瘍細胞が均一に増殖。

GFAPに陽性を示す好酸性の細胞質と偏在する核を有する。

腫瘍はIDH1,Olig2に陽性でATRX発現が保持されている。
弱拡大では,多分岐する毛細血管網とそれらと無関係にびまん性に分布する小型円形細胞の均一な増殖がみられる(Figure 13.70)。腫瘍細胞は細胞質および線維性突起に乏しく,星細胞腫でみられる血管周囲および背景のglio-fibrillary backgroundは目立たない(Figure 13.71)。核は正円形で,ほぼ裸核状で高いN/Cを示す。微細顆粒状のクロマチンパターンを呈し,小型の核小体を認めるが非常に単調な形態を示す(Figure 13.72)。退形成性乏突起膠腫では,明らかに細胞密度が高く,内皮細胞が腫大・重層化し,多分岐した毛細血管の増生を認める。核分裂像も散見され,核形不整や大小不同も増加する。乏突起膠腫に比べてminigemstocytesの出現頻度が高い。

毛細血管周囲に集簇して分布する星細胞系腫瘍と異なり,本腫瘍では小型円形細胞が血管と無関係にびまん性に分布する。血管周囲の網目状の線維性背景に乏しい。

腫瘍細胞は小型かつ均一な所見。一部に好酸性の細胞質を有する細胞も認めるが,線維性の細胞質突起は目立たない。

正円形の核と明るい細胞質を有する単調な腫瘍細胞。核網は星細胞系腫瘍と比較してやや微細~粗顆粒状を示す。
鑑別診断として悪性リンパ腫,転移性脳腫瘍,星細胞腫,小型細胞よりなる膠芽腫が挙げられるが,核の切れ込みなどの形状不整,出現細胞の多彩性,細胞質突起の有無ならびに毛細血管との関係性などが重要な観察ポイントである3),30),31)。
4. 毛様細胞性星細胞腫(pilocytic astrocytoma) 1) 概要星細胞系腫瘍群の中で比較的限局した腫瘤を形成し,特有の臨床像および遺伝子異常を示す腫瘍群は「限局性星細胞腫瘍」として区別されており,その代表的な腫瘍が毛様細胞性星細胞腫である。本腫瘍は双極性の細長い細胞突起を伸長する星細胞(piloid cell)が充実性に増生する部分と微小嚢胞を伴った水腫様の背景に疎に増生する部分からなる二相性(biphasic pattern)を特徴とする。小児~若年成人に多く,15歳未満が半数を占める。小脳,脳幹,視神経,視床下部に好発し,視神経・視交叉発生星細胞腫の大部分が本腫瘍である。大脳半球にも発生するがその年齢層はより高い。全摘出術により完治が可能な良性腫瘍で予後は良好である(WHO grade I)23)~27)。
2) 画像・肉眼所見(Figure 13.73)
T1強調で低信号(左),造影MRIで強く増強される。肉眼的には限局性の軟らかい腫瘤(黄矢印:腫瘍,青矢印:正常視神経)。
境界明瞭な嚢胞性腫瘤として描出されることが多く,嚢胞壁に結節性病変を伴う。T1強調で低信号,T2強調で高信号を示し,ガドリニウムで強く増強される。肉眼的には限局性で軟らかく,嚢胞を伴うことが多い。
3) 遺伝子学的所見本腫瘍の約70%の症例(特に小脳発生)でKIAA1549-BRAF融合遺伝子が認められる。これは両遺伝子が近接して存在する7q34領域における縦列重複によるものである。この融合遺伝子はBRAFのN末端側がKIAA1549のN末端側に置き替えられたもので,転写産物はBRAFのリン酸化酵素活性が恒常的に亢進したがん遺伝子活性を有する。本腫瘍を最も特徴づける所見であり,RT-PCR法あるいはBRAF遺伝子のbreak apart probeを用いたFISH法が有用である。その他の遺伝子異常として,NF1変異や5%程度の症例でBRAF(V600E)変異がみられることが知られている。IDH変異は認められない23)~27)。
4) 組織像(Figure 13.74)
左:充実性領域と疎な海綿状領域との二相性組織像が明瞭。
右:細長い細胞質突起と細胞間にRosenthal線維がみられる。
発生部位や症例によってその量的比率の差異はあるが,基本的には充実性領域と疎な海綿状領域との二相性が明瞭である。腫瘍細胞は小型かつ単調で,毛髪様の細長い細胞質突起を有する。充実性領域では腫瘍細胞の密な束状配列がみられ,線維間には棍棒状あるいは不定形の好酸性を示す小体で,星細胞突起の変性構造物であるRosenthal線維が観察される。海綿状領域では浮腫状から微小嚢胞性の背景に腫瘍細胞がまだらに分布するため,双極性の細胞質突起がより明瞭である。しかし,核の腫大,クロマチン増加,大小不同などの多形性がより目立つ傾向にある。またこの領域ではRosenthal線維と同じく星細胞突起の変性構造物で,球状~類円形を呈し,エオジン好染性,PAS反応陽性の顆粒を含む好酸性顆粒小体(eosinophilic granular body; EGB)がみられる。
本腫瘍は間質の毛細血管網が豊富で密な増生を伴うため,退形成性星細胞腫や膠芽腫との鑑別が問題となることがある。細胞質突起の性状,血管内皮細胞の核腫大や多層化,核分裂像の有無に加えて,上記2つの星細胞突起の変性構造物出現の有無が重要な鑑別点である23)~27)。
5) 圧挫細胞像組織所見同様,圧挫標本でも二相性が特徴的である。細長い線維性突起が明瞭な細胞が平行に並び束状集塊を形成する密な部分と,網目状の線維性背景がより目立つ疎な部分が観察される(Figure 13.75)。腫瘍細胞は放射状に突起を伸長する細胞も混在するが基本的には双極性(二方向性)の細長い線維を伸長し,細胞質はほとんどみられない。核所見は極めて単調で,楕円形~紡錘形を示し,繊細な核クロマチン網を呈する(Figure 13.76)。圧挫標本では凍結標本に比べて明らかにpiloid cellの認識が容易で,またRosenthal線維および好酸性顆粒小体も明瞭に観察が可能なため本腫瘍の診断には圧挫標本の併用が極めて有効である(Figure 13.77)3),30)~32)。
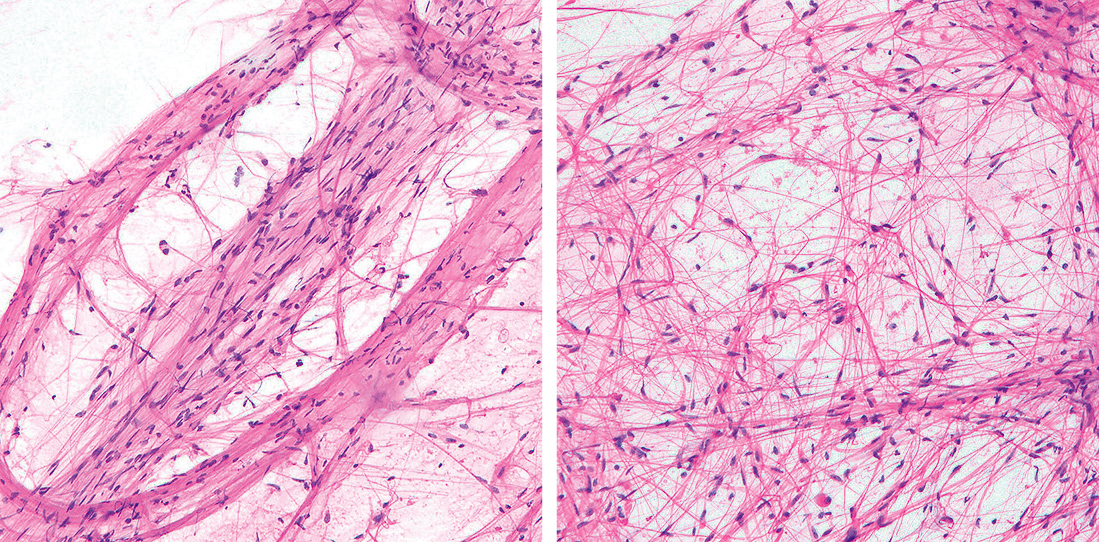
細胞が密な束状集塊(左)と疎な領域(右)の二相性が明瞭。

繊細で毛髪様の細長く伸びた双極性の細胞質突起が明瞭。

左:好酸性で太い棍棒状を呈するRosenthal線維。
右:好酸性顆粒状の類円形を示すeosinophilic granular body。
本亜型は,乳幼児の視床下部・視交叉部に好発し,局所再発や髄腔播種を起こし易く,相対的に悪性度が高い亜型とされている(Figure 13.78)。組織学的には粘液様基質を背景にpiloid cellの単調な増殖を認める。二相性がみられず血管周囲性偽ロゼット構造あるいは血管中心性配列(angiocentric pattern)と呼ばれる特徴的な構築を示す(Figure 13.79)。またRosenthal線維やEGBが認められないのも特徴である。壊死や脳実質浸潤がみられ,Ki-67標識率は2~20%(平均4%)と明らかに高い23)~27)。

6歳男児。視床下部発生のPMAで広範に脳実質に浸潤し,第3脳室壁を穿破し,髄腔播種をきたした症例。

粘液水腫様基質を伴い細長い繊細な突起をもつpiloid cellが血管周囲に偽ロゼット状に配列する。二相性はみられない。
圧挫標本では,組織と同様に血管中心性配列が明瞭に観察され,ヘマトキシリンに淡染する粘液浮腫状の背景に,極めて単調な形態を示すpiloid cellがみられる。血管周囲では腫瘍細胞が柵状に整列し,細長い線維性突起を伸長する像が目立つ。通常の毛様細胞性星細胞腫と比べて,全体的に均一で,多彩性に欠き,核はより正円形で核分裂像の出現頻度が高い(Figure 13.80)3),30)~32)。

血管周囲性に集簇し,核が柵状に整列する偽ロゼット構造が明瞭(左)。粘液腫様の背景に類円形核と繊細な細胞質突起を有する腫瘍細胞。核は単調で多形性に欠く(右)。
「限局性星細胞腫瘍」の一つで神経細胞にも類似する肥胖細胞様の大型星細胞よりなる腫瘍で結節性硬化症(tuberous sclerosis; TS)との関連が深い。TSの10~20%に発生するが,TSと関連のない症例もある。小児から若年成人に好発する(WHO grade I)。TSの病因遺伝子は9p34のTSC1遺伝子と16p13.3のTSC2遺伝子で,いずれもがん抑制遺伝子であるが,両遺伝子異常による機能喪失がTS発症の原因である23)~29)。
2) 画像所見(Figure 13.81)
腫瘤は側脳室壁のMonro孔付近に好発し,T1強調で低信号(左),T2強調で高信号(右),造影MRIで強く増強される。
90%以上の症例が側脳室に発生し,Monro孔周囲の脳室壁に境界明瞭な結節性病変を形成する。嚢胞を伴うことが多い。MRI-T1強調で低信号,T2強調では不均一な高信号を示し,ガドリニウムで強く造影される。CTでは石灰化がみられることが多い。
3) 組織像(Figure 13.82)
拡張した血管が豊富な腫瘍で,大型核と好酸性の豊富な細胞質を有する星細胞が増殖。核小体が目立つ細胞も目立つ。
肥胖性星細胞様の大型細胞と線維性細胞突起をもつ紡錘形細胞よりなる。広い好酸性の細胞質を有する大型細胞の核網は淡く繊細で,単個の大型核小体を有するため神経細胞に類似する。紡錘形細胞は小型で流れるような束状配列を示す。間質には拡張した血管が豊富で海綿状血管腫に見えることもある。本腫瘍は星細胞系腫瘍に分類されるが,膠細胞系と神経細胞系両者の免疫形質を示す。すなわちGFAP,S100蛋白,nestinに陽性を示すと共に,種々の程度で神経細胞分化を示唆するneuron-associated class IIIβ-tubulin,Neu-N,neurofilament蛋白に陽性を示す。
4) 圧挫細胞像(Figure 13.83, 13.84)
突起を有する紡錘形細胞と肥胖星細胞様の大型細胞が混在。
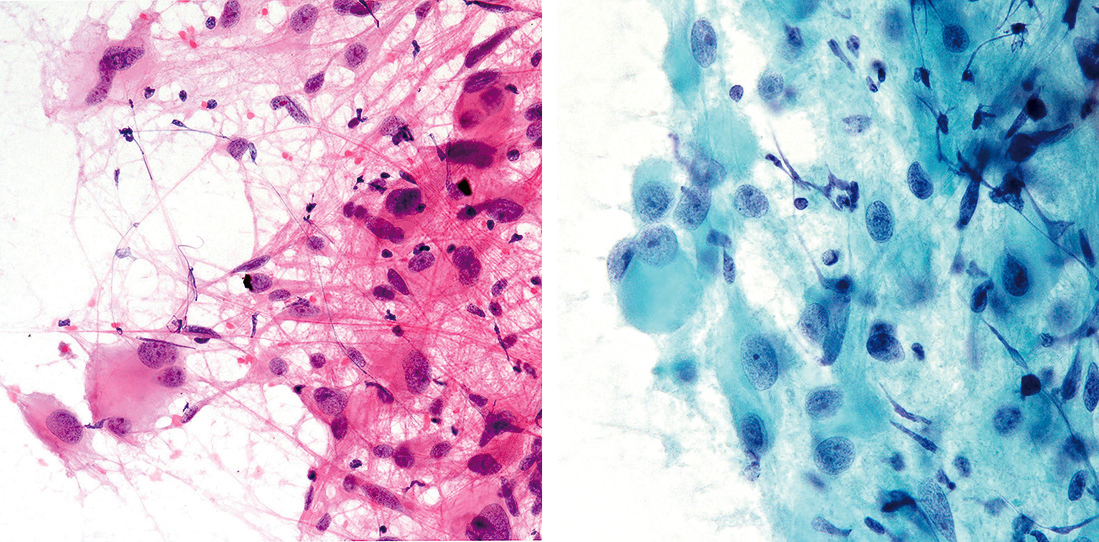
大型細胞は繊細な核網と核小体が明瞭で神経細胞に類似。
びまん性星細胞腫および毛様細胞性星細胞腫に類似した線維性突起が豊富な紡錘形細胞の増殖に加え,肥胖細胞類似の大型細胞を認める。小型紡錘形細胞は細長い細胞質体を有するためstrap cellと表現されている。大型細胞の核網は繊細で,核小体が目立ち,細胞質は広く一部で顆粒状を示す。出現する細胞に大小不同性など多形性が強いため,膠芽腫を含めた他の星細胞系腫瘍との鑑別が問題となるが,患者年齢とMonro孔近傍の側脳室壁に局在する腫瘍,さらに神経細胞類似の大型細胞の混在という所見から本腫瘍の診断は比較的容易と思われる3),33)。
6. 上衣腫(ependymoma) 1) 概要上衣細胞への分化を示す小型かつ均一な細胞からなり,脳室壁や脊髄に境界明瞭な腫瘤を形成する。上衣腫の約60%が後頭蓋窩に発生し,30%がテント上,10%が脊髄に発生する。後頭蓋窩のほとんどが第四脳室壁(特に底部)に好発し,多くが小児でみられる。テント上では側脳室ならびに第三脳室壁に多くが発生するが,脳室と離れた脳実質に発生することもある。脊髄発生症例は成人に多い23)~27)。
2) 画像および肉眼所見脳室壁に連続性のある境界明瞭な腫瘤で,MRI-T1強調で低信号,T2強調で不均一な高信号を示す。一般的にガドリニウムで強く増強される(Figure 13.85)。肉眼的には淡褐色調で軟らかく,周囲の脳実質との境界は明瞭で,出血・壊死は目立たない。

不均一に造影される第4脳室壁から発生した巨大な腫瘤。
腫瘍細胞は均一な類円形の核で微細な顆粒状クロマチン網を呈し,細胞質は淡い好酸性または淡明で個々の細胞境界は不明瞭である。線維性の細胞質突起が明瞭な細胞も混在する。上衣腫に特徴的な組織構築として二つの細胞配列パターンがある。一つは血管周囲性偽ロゼットと呼ばれ,腫瘍細胞が血管周囲に集簇し,細長い線維性突起を血管に伸長して花輪状に配列する構造である。血管近傍は線維のみで核が乏しい帯状の領域を形成し,無核帯と呼ばれる(Figure 13.86)。もう一つが真の管腔を取り囲むように腫瘍細胞が花冠状に配列する真性ロゼットである。内腔面は直線的で,管腔のサイズが小さいものから上衣ロゼット,上衣細管および上衣管と呼ばれる(Figure 13.87)。免疫染色にて腫瘍細胞はGFAPに陽性を示すが,細胞体より突起での発現が強いため血管周囲偽ロゼット部分でより強い陽性を示す。またEMAの細胞間および細胞内のドット状あるいは小リング状の陽性所見は本腫瘍診断に特異度の高い重要な所見である(Figure 13.88)23)~29)。

血管周囲に腫瘍細胞が花輪状に配列し,血管近傍は細胞突起よりなる無核帯が形成される。

左:上衣管(ependymal canal),右:上衣細管(ependymal tubule)。

GFAPは細胞突起に陽性。上衣管内腔に加え,細胞間および細胞内にみられるEMAのドット状や小リング状の陽性像。
形態学的亜型として,乳頭状の細胞配列が目立つ乳頭状上衣腫,乏突起膠腫に類似した淡明な細胞質を有する明細胞上衣腫,上衣ロゼットや血管周囲性偽ロゼットの形成が乏しく,繊細な突起を伸ばす双極性細胞が束状配列する伸長細胞性上衣腫がある。中でも明細胞上衣腫は若年者のテント上に好発し,分枝毛細血管がよく発達した明細胞血管亜型は近年の遺伝子研究で,RELA融合遺伝子陽性の上衣腫に該当することが判明している。RELA遺伝子は炎症や細胞増殖,アポトーシスなどに関与する転写因子であるNF-κBを構成する蛋白の一つをコードするが,融合遺伝子によってNF-κBシグナル系が活性化され腫瘍が発生するとされている。本亜型は上衣腫の中でも相対的に予後不良との報告がある23),26),27)。
4) 電子顕微鏡所見(Figure 13.89)
細胞間および細胞質内の小腺腔(赤矢印)には多数の微絨毛(青矢印)が認められ,細胞間にはジッパー様の接着装置 (黄矢印)が発達している。
腫瘍細胞間には線毛や微絨毛が発達し,gap junctionなどの接着構造が観察される。細胞質内にも小腺腔を認めることがあり,内腔面にも多数の微絨毛が発達している。これらはalcian blue染色に陽性を示し,免疫染色におけるEMAのリング状およびドット状陽性所見を示す部位に一致している23)~29)。
5) 圧挫細胞像組織所見と同じく,血管周囲性偽ロゼット構造が最大の特徴で,血管周囲では繊細な線維性突起が認められ,無核帯も明瞭に観察される(Figure 13.90)。毛細血管の分枝により偽乳頭状の集塊を形成することが多い。個々の腫瘍細胞は均一な所見で,核は小型・類円形で微細~粗顆粒状のクロマチンが均等に分布する。小型の核小体を1~2個認めることが多い。集塊の中心部に向かって線維を伸長し,時に明瞭な管腔を伴うロゼット構造がみられる(Figure 13.91)。時にalcian blue陽性の細胞質内空胞と偏在核を有する印環細胞様の形態が目立つ症例や,微絨毛や線毛が発達しているため集塊辺縁に毛羽立ちを認める重積性のある集塊がみられるため,転移性腺癌との鑑別が問題となる症例もある。核異型などの細胞の多形性を評価することが重要である(Figure 13.92)。

血管周囲に腫瘍細胞が集簇し,偽乳頭状構造を呈する。圧挫標本でも無核帯の存在が確認できる。

内腔側に線維性突起を伸長する上衣管(ロゼット)構造。核は小型・類円形で多形性に乏しく均一な所見を示す。

左:印環細胞様の形態を示す上衣腫細胞。
右:微絨毛に相当する集塊辺縁部での細胞質の毛羽立ち。
鑑別すべき脳室内に好発する腫瘍として,テント上では上衣下巨細胞性星細胞腫,中枢性神経細胞腫,脈絡叢乳頭腫,脳室内に進展する星細胞系腫瘍が挙げられるが,いずれも上衣腫の特徴的な細胞所見から鑑別は容易である。しかし乳幼児・小児の第四脳室発生で退形成性変化が強い症例(退形成性上衣腫)の場合,髄芽腫との鑑別が問題となることがある。神経細線維性の基質(ニューロピル)の有無が重要な鑑別点となる(髄芽腫の項参照)3),30),34)。
7. 脈絡叢乳頭腫(choroid plexus papilloma) 1) 概要脈絡叢上皮細胞に由来する稀な腫瘍で,異型細胞が著明な乳頭状構造を作りながら増殖する。退形成性の程度より,脈絡叢乳頭種,異型脈絡叢乳頭種,脈絡叢癌の三型に分けられるが,90%近くが脈絡叢乳頭種である。小児に好発し,約半数の症例が15歳未満である。約80%が側脳室に発生し,次いで第四脳室(12%),第三脳室(8%)が好発部位である。成人ではその多くが第四脳室に発生する23)~29)。
2) 画像および肉眼所見(Figure 13.93)
左:造影MRI(T1強調)。均一に強く増強される側脳室腫瘤。
右:肉眼像。黄褐色顆粒状でカリフラワー状を呈する。
MRI-T1強調で等信号,T2強調で高信号,造影剤で強く増強される。肉眼的には脳室壁に付着した黄褐色顆粒状の境界明瞭な腫瘍で,しばしば「カリフラワー状」と形容される。
3) 組織像および免疫学的所見(Figure 13.94, 13.95)
線維血管性の間質を軸に樹枝状に分岐し,乳頭状に増殖。

表面は均一かつ異型の弱い核を有する立方状の細胞が単層に整列し増殖。腫瘍細胞はtransthyretinに強陽性を示す。
単層の円柱状あるいは立方状の上皮細胞が細い線維血管性の軸を伴い乳頭状に増殖する。細胞表面はコロイド鉄染色およびEMA陽性の線毛・微絨毛を有し,直線的である。核は基底側に整列し,極性の乱れはなく均一で異型に乏しい。腫瘍細胞の細胞質が好酸性顆粒状のoncocytic変化を示すこともある。異型脈絡叢乳頭種(WHO grade II)は,「高倍率10視野で2個以上の核分裂像を認める」診断基準がある。脈絡叢癌(WHO grade III)では,明らかな悪性像を示し,小児に好発するが,乳頭状構造が不明瞭になり,細胞密度が高い充実性増殖を示し,壊死を伴う。免疫学的にtransthyretin(prealbumin),synaptophysinに陽性,一部の細胞でGFAP陽性を示す。一般的にはCK7陽性 CK20陰性のパターンを示す。WHO gradeが上がるほど上記マーカーの陽性率は低くなる23)~27)。
4) 圧挫細胞像(Figure 13.96)
乳頭状集塊を認めるが,単層で核の極性は保持されている。
線維血管性の軸を有する著明な乳頭状集塊を認める。核は小型かつ均一で,集塊は単層のシート状を呈し,重積性に乏しい。集塊辺縁部は直線的で核は整列し,配列の乱れを認めない。細胞表面は発達した線毛を反映して毛羽立ち感がある。鑑別診断として小児では乳頭状上衣腫が挙げられるが,線維性突起や血管周囲性偽ロゼットの有無が重要で,成人では転移性腺癌が挙げられるが,核異型が異なる。
8. 中枢性神経細胞腫(central neurocytoma) 1) 概要神経細胞系腫瘍の中でも代表的な腫瘍で側脳室のMonro孔付近に好発する。20~30歳台の若年成人に多く,増殖能は低い(WHO grade II)。同じ組織像と免疫形質を持つ脳実質発生の腫瘍は脳室外神経細胞腫(extraventricular neurocytoma)とされる23)。
2) 画像および肉眼所見(Figure 13.97)
T1強調で等~低信号,造影MRIで不均一に増強される。
T2強調では多嚢胞性で濃淡不均一な信号を示すことが多い。
T1強調で等~低信号,T2強調では多嚢胞性で,造影MRIで不均一に増強される。CTでは石灰化像を認めることが多い。肉眼的には脳室壁に付着する灰白色調の軟らかい腫瘤を形成する。
3) 組織像および免疫学的所見類円形核と淡明な細胞質をもつ極めて単調な小型円形細胞が敷石状に増殖する。核周囲明庭が目立つこともあり,極めて乏突起膠腫に類似している。細胞間には微細顆粒状を示す神経細線維性の基質(ニューロピル)が豊富で,これらを囲むように腫瘍細胞がロゼット様に配列する像や,豊富な細胞質と明瞭な核小体を持つganglion cellを認めることもある(Figure 13.98)。約半数の症例で石灰化小体が観察される。腫瘍細胞は神経細胞系マーカーのNeu-N,MAP2などに陽性,Olig2には陰性を示す(乏突起膠腫との鑑別点)。細胞質および背景の微細顆粒状を呈するニューロピルはsynaptophysinに強陽性を示すが,neurofilament proteinとchromogranin Aは陰性のことが多い23)~29)。

類円形核と淡明な細胞質を有する細胞が単調に増殖。細胞間には細線維性基質を認め,ロゼット配列もみられる。
類円形で単調な腫瘍細胞が孤在性あるいは平面的な小集塊を形成して出現する。核はほぼ正円形で,核クロマチンは微細~粗細顆粒状で均等分布し,小型の核小体を数個認める。細胞質は細顆粒状を呈するが個々の細胞境界は不明瞭で裸核細胞も目立つ(Figure 13.99)。背景には細胞質と同様のエオジンおよびライト緑好染性の微細顆粒状物質を認め,これはsynaptophysinに陽性を示す(Figure 13.100)。鑑別診断として,形態的には乏突起膠腫や上衣腫が挙げられるが,ニューロピルの認識が本腫瘍を推定する最も重要な所見である。患者年齢と発生部位,脳実質との連続性は不可欠な臨床情報である3),30),34)。

類円形で単調な腫瘍細胞が構築に乏しくびまん性に出現。

細胞質と背景の微細顆粒状の基質はsynaptophysinに陽性。
小児悪性脳腫瘍として最も頻度の高い高悪性度の脳腫瘍。乳幼児・小児の小脳および第4脳室近傍に発生し,未熟な神経上皮性細胞が密に増殖する。概して予後は不良で約85%の症例が15歳未満に発生する。増殖・浸潤能が著しく高く,第4脳室内を占拠し,閉塞性水頭症や種々の頭蓋内圧亢進症状を示す。2016年WHO分類より大幅な改定がなされ,従来の組織学的定義に加えて,分子遺伝学的定義が設けられた。組織学的には,古典的髄芽腫(classic),線維形成性・結節性髄芽腫(desmoplastic/nodular),高度結節性髄芽腫(~with extensive nodularity),大細胞・退形成性髄芽腫(large cell/anaplastic)に分類される。分子遺伝学的には,Wnt伝達経路の活性化を示すWNT-activate,Sonic Hedgehog(SHH)経路に属する遺伝子群が活性化したSHH-activated,特異的なシグナル伝達経路の関与が未だ判明していないnon-WNT/non-SHHの3群に大別され,更にSHH-activatedはTP53-mutantとTP53-wildに,non-WNT/non-SHHはgroup 3とgroup 4にそれぞれ分類される。各々の亜型によって好発年齢,予後,発生母地ならびに組織像が異なる。組織学的および分子遺伝学的定義の相互関係をFigure 13.101に示す23)~27)。

小脳虫部~第4脳室背側を主座にMRI-T1強調で低信号,T2強調で高信号を呈し,ガドリニウムで不均一に強く増強される腫瘤を形成する。早期の段階から髄腔播種を伴っていることが多い(Figure 13.102)。高度結節性髄芽腫は小脳半球に多く,造影MRIで「ブドウの房状」と称される特徴的な画像を示す。肉眼的には淡桃色~灰白色で軟らかい性状を示す。

第4脳室内を占拠する不均一に強く増強される腫瘤。
N/Cが高い小型細胞が充実性,髄様に増殖する。神経細胞への分化能が高い症例では,細胞間に微細顆粒状を示す神経細線維性の基質(ニューロピル)が豊富で,その線維性突起を伸ばす腫瘍細胞の花冠状配列を認める。これはHomer-Wrightロゼットと呼ばれ,髄芽腫の特徴である(Figure 13.103)。核は円形~卵円形で,粗顆粒状のクロマチンが密に増量し,多くの核分裂像やアポトーシス像がみられる。古典的髄芽腫は髄芽腫全体の約70%を占める。分子遺伝学的にはnon-WNT/non-SHH型が最も多いが,WNT-activated型の大部分も古典的髄芽腫としての組織像を示す。WNT-activated亜型の古典的髄芽腫は最も予後良好で,5年生存率は95%にも達する26),27)。

中心部の細線維性基質よりなる無核帯を囲み小型異型細胞が花冠状に並ぶHomer Wright型ロゼットがみられる。
豊富な好銀線維網が形成された小型髄芽腫細胞が密に増殖する部分と島状の境界明瞭な淡明領域「pale island」が混在する亜型。pale island内には好銀線維が認められず,細胞密度も低いため相対的に明るく見える(Figure 13.104)。結節内の細胞は神経系への分化傾向が強く,synaptophysin,Neu-N,MAP2などが高発現する一方,増殖活性が低いためKi-67標識率は低い(Figure 13.105)。本亜型は髄芽腫全体の約20%を占め,小脳半球の表層部に腫瘤を形成する。分子遺伝学的には,ほぼ全例がSHH-activated TP53-wild typeに相当し,3歳以下の乳幼児に発生する髄芽腫の約半数以上がこの亜型である。WNT-activated亜型に次いで予後良好で,5年生存率は約80%である26),27)。

境界明瞭な淡明領域“pale island”を認める。周囲では豊富な好銀線維網が形成されるが,結節内では線維を認めない。

Pale island内の細胞密度は低く,核網が繊細で,淡い細胞質を有する細胞が集簇する。細線維性基質が豊富で神経細胞系のマーカーが高発現し,この領域での細胞増殖能は低い。
組織学的に線維形成性・結節性髄芽腫と同類であるが,pale islandがより大型で融合し,結節間の暗調な領域が狭い。結節内では神経細線維性の基質が豊富で,流れるような数珠状の配列を示す小型の神経細胞に類似した腫瘍細胞が増殖する。これらの細胞の核は中枢性神経細胞腫の核所見に類似した粗大~微細顆粒状のクロマチン網を呈し,小型の核小体を有する。(Figure 13.106)。分子遺伝学的には,線維形成性・結節性髄芽腫と同様,全例がSHH-activated TP53-wild typeに相当し,予後は良好である。本亜型は従来,小脳神経芽腫(cerebellar neuroblastoma)と呼ばれていた腫瘍に相当するものである23)~29)。

線維形成結節性髄芽腫より大型のpale islandが目立つ。結節内では細線維性基質が豊富で,流れるように配列する。
大型の均一な核を有する腫瘍細胞からなる髄芽腫を大細胞型と呼び,多形性が顕著で明らかに多数の核分裂像やアポトーシス像,腫瘍細胞が他の腫瘍細胞を包み込む像(cell wrapping)や鋳型像(molding)が目立つ亜型を退形成型と呼称されていたが,両者は混在することが多いため,2016年WHO分類からは一つの亜型として統合された。分子遺伝学的には,多くがnon-WNT/non-SHH(特にgroup 3)またはSHH-activated TP53-mutant typeである。乳幼児および小児に多く,髄芽腫の中で最も予後の悪い亜型である23)~27)。
4) 細胞像細胞診標本ではほぼ裸核状の腫瘍細胞がびまん性に出現する。核クロマチンは微細~粗大顆粒状で,核小体は目立たない(Figure 13.107)。また人参様“carrot-shaped”と形容される一端が尖った短紡錘形の核を有する細胞もみられる(Figure 13.108)。線維形成性・結節性および高度結節性髄芽腫でよくみられる所見であるが,神経細胞系への分化が著明な症例では,中枢性神経細胞腫と同様の好酸性で顆粒状の細胞質および細胞外基質が目立ち,それらを囲むような腫瘍細胞のロゼット配列や流れるような配列を認める。またこれらの細胞は周囲の髄芽腫細胞に比べて,若干大きく,核膜が菲薄で,非常に繊細で明るいクロマチン網を呈し,単個の明瞭な核小体を有する(Figure 13.109)3),30),34)。

核クロマチンは細顆粒状で均等分布。核小体は目立たない。

核は楕円~正円形を示し,人参様(carrot-shaped)と称される一端が尖った紡錘形の核が混在する。

細線維性基質に相当する微細顆粒状物質とそれを囲むように腫瘍細胞が配列するロゼット構造がみられる。
髄芽腫では様々な程度でneuron specific enolase,MAP2,Neu-N,synaptophysinやClass IIIβ-tubulinなどの神経細胞系マーカーが陽性となるが,これらは小型細胞が密に増殖する部分よりpale island内やHomer-Wrightロゼット部分で強陽性を示す。膠細胞(glial cell)への分化を示唆するGFAPにも陽性を示す細胞も混在するがその陽性率は10%程度と低い(Figure 13.110)。分子遺伝学的分類に有効な免疫マーカーも幾つか報告されている。WNT-activated亜型ではWnt伝達経路の一つであるCTNNB1遺伝子(β-catenin)の変異を認めるため,β-cateninが核内に発現する。他のSHH-activatedおよびnon-WNT/non-SHH亜型では細胞膜に陽性となるため鑑別に有用である。GAB1もまた有効な鑑別マーカーで,SHH-activated亜型だけが細胞質内に陽性を示す。特に線維形成性・結節性髄芽腫および高度結節性髄芽腫での結節間の暗調な増殖部分に強陽性を示す(Figure 13.111)。YAPはWNT-activated亜型とSHH-activated亜型の両方で,核および細胞質に陽性所見を示すが,non-WNT/non-SHH亜型では陰性となるため,相対的に予後不良群であるgroup 3とgroup 4の抽出に有効である23),26),27)。

GFAP陽性の腫瘍細胞が少数混在し,細胞間の細線維性基質はsynaptophysinに陽性を示す。

線維形成結節性髄芽腫。SHH-activated型ではYAPに核と細胞質が陽性,GAB1が細胞質に陽性を示す。
末梢神経の髄鞘形成細胞であるシュワン(schwann)細胞に由来する良性腫瘍(WHO grade I)。30~60歳台に多く,小児では稀である。女性は男性より1.5倍ほど多い。頭蓋内においては第VIII脳神経(前庭神経)発生が多く約90%以上を占める。三叉神経,顔面神経などからも発生するがその頻度は低い。脊髄神経発生は感覚神経である後根から生じるものが多い。前庭神経は脳幹から10~13 mm程度までは乏突起膠細胞由来の中枢性髄鞘に覆われているが,内耳孔内ではschwann細胞由来の末梢性髄鞘に移行する。本腫瘍はこの移行部から発生することが知られており,第VIII脳神経のシュワン細胞腫は前庭神経節近傍から発生し,小脳橋角部に向かって進展する。従来「神経鞘腫」の名称が使用されていたが,神経鞘が髄鞘を指す言葉であり,シュワン細胞由来の腫瘍という観点から2016年のWHO分類からこの名称は使用されなくなった23)~27)。
2) 画像および肉眼所見MRI-T1強調で低信号,T2強調で高信号を呈し,ガドリニウムで均一に増強される。肉眼的には被膜を有する表面平滑で境界明瞭な腫瘤を形成する。割面は淡黄色~淡褐色で,ゴム様の硬度で嚢胞や出血を伴う(Figure 13.112)。

右小脳橋角部に均一に増強される腫瘤を認める。腫瘍は淡黄色~淡褐色で,ゴム様の硬度で嚢胞や出血を伴う。
紡錘形の腫瘍細胞が線維束を作り密に並ぶ部分と,細胞密度が低く水腫様の基質を伴い網目状に増殖する部分との二相性(biphasic pattern)が認められる(Figure 13.113)。前者の密な部分はAntoni A領域,後者の疎な部分はAntoni B領域と呼ばれる(Figure 13.114)。腫瘍細胞の核は細長く,両端が尖り,クロマチン増量に乏しく,均一な所見を示す。細胞質は淡い好酸性を示し,合胞状で個々の細胞境界は不明瞭。細胞の両端からは細長い線維性突起が観察される。Antoni A領域での核は特に細長く,葉巻状と形容される。この領域では核が横一列に並ぶ柵状配列がみられ,この所見は脊髄発生症例でより目立つ。また柵状配列が近接して存在し,両者間に線維性突起のみから構成される無核帯が存在し,ヴェロケイ小体(Verocay body)と呼ばれる(Figure 13.115)。

細胞密度に疎密がある二相性パターンを示す。

細胞が線維束を作り密に並ぶAntoni A領域(左)と水腫様の基質を背景に網目状に分布するAntoni B領域(右)。

核の柵状配列(左)と無核の帯状領域(Verocay body:右)。
Antoni B領域での腫瘍細胞の核はAntoni A領域と比較して,類円形で丸みがあり,大小不同やクロマチンの濃染,形状不整などの多形性が強い。この領域では腫瘍細胞が線維性突起を有する様がよく分かる。またうっ血,血栓形成や壁の硝子化を伴った毛細血管の増生,ヘモジデリンの沈着およびそれらを貪食した泡沫組織球の浸潤,大小の多房性嚢胞の形成もしばしば観察される23)~29)。
4) 免疫学的所見および遺伝子異常免疫染色にて腫瘍細胞は,S100蛋白,vimentinに強陽性を示す。GFAP,EMAは基本的に陰性を示す。シュワン細胞腫の約60%でNF2遺伝子変異がみられる。両方のアレルでの変異・欠失により転写産物であるmerlinの機能が喪失し,増殖シグナルの制御異常やアポトーシス抑制が起こり,腫瘍が発生すると考えられている26),27)。
5) 細胞像腫瘍組織が硬く,結合性が強い紡錘形細胞の束状集塊が主体となる腫瘍であるため,圧挫標本では引き伸ばされ難く,また捺印標本に孤在性の腫瘍細胞がほぼ付着しないという所見が本腫瘍の特徴である。圧挫標本においては,錯綜する細胞密度の高い紡錘形細胞の束状集塊と,網目状の線維性細胞突起が疎に分布する二相性がみられる(Figure 13.116)。密な束状集塊,すなわちAntoni A部分では両端が尖った長楕円形~紡錘形で,クロマチンが繊細・顆粒状を呈する核の平行に配列する像がみられ,核膜の肥厚や肥大した核小体はみられない。繊細で細長い細胞質突起を認める(Figure 13.117左,13.118)。一方,細胞密度が疎なAntoni B部分では細長い線維性突起による網目状構造が明瞭で核の大小不同,クロマチン粗大化や巨核細胞などを認めるため,他の腫瘍との鑑別が問題となる事がある(Figure 13.117右)。特徴的な配列を示すAntoni A領域の認識と腫瘍部位が診断の重要なカギである。鑑別診断として最も重要なのが髄膜腫であるが,本腫瘍と比べて細胞の結合性が弱く,次に挙げる所見が異なる(Figure 13.119)。髄膜腫では,1)捺印標本中に数多くの孤在細胞および小集塊が出現。2)集塊辺縁部での細胞のほつれが強い。3)錯綜する束状集塊が互いに融合し,その部分で丸みのある腫大した核が目立つ30),35)。

錯綜する細胞密度の高い束状集塊と網目状の線維性の細胞突起が疎に分布する部分との二相性がみられる。

Antoni A部分での核は紡錘形で平行に並び(左),Antoni B部分での核は楕円~類円形で丸みがあり大小不同を伴う。しかし双極性の長い突起を有する様子がよく分かる(右)。

核は両端が尖った長楕円形~紡錘形で,クロマチンは繊細な顆粒状を呈する。繊細で細長い細胞質突起を認める。
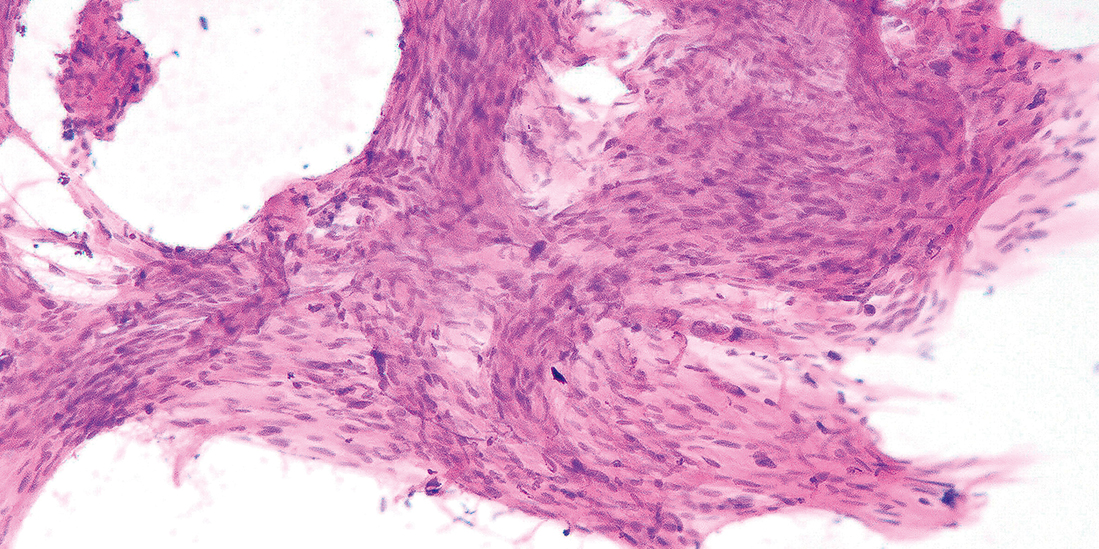
シュワン細胞腫では,集塊辺縁が平滑で,細胞のほつれがなく,錯綜する束状集塊は折り重なるだけで融合しない。
髄膜皮細胞あるいはクモ膜細胞より発生する腫瘍で,その発生頻度は高く,原発性脳腫瘍の約1/4を占める。30~70歳台の成人に多く,乳幼児・小児では稀である。男女比に関しては女性に多い(約2.7倍)が,悪性度が高い亜型は男性に多い。組織像は多彩で2016年のWHO分類では組織像と悪性度に基づいて以下の3群に分類される23)~27)。
・低異型度髄膜腫(low grade, WHO grade I)
髄膜皮性髄膜腫,線維性髄膜腫,移行性髄膜腫に代表され,その他に6組織型がある。最も頻度が高く,完全切除ができれば再発リスクは低く,予後は良好。
・中間異型度髄膜腫(intermediate grade, WHO grade II)
異型髄膜腫(低異型度の脳浸潤を含む),脊索腫様髄膜腫,明細胞髄膜腫が含まれ,再発率がやや高い。
・高異型度髄膜腫(high grade, WHO grade III)
退形成性髄膜腫,乳頭状髄膜腫,ラブドイド髄膜腫が含まれ,浸潤能が高く,再発・転移のリスクが高い。
本項では,日常遭遇する機会の多い亜型を中心に各々の特徴的な組織・細胞像について概説する。
2) 画像および肉眼所見(Figure 13.120)
均一に増強される腫瘤で近接する硬膜が同時に造影される“dural tail sign”が特徴的(赤矢印)。硬膜(黄矢印)。
MRI-T1強調T2強調共に,等信号のことが多く,ガドリニウムで均一に強く増強される。連続する硬膜が同時に造影される“dural tail sign”が特徴的。肉眼的に黄白色調で弾性硬の充実性腫瘤で,周囲との境界は明瞭である。亜型によっては,嚢胞や石灰化を伴うもの,周囲脳に著明な浮腫を伴うものがある。高異型度髄膜腫では,腫瘍辺縁が不整形で凹凸を伴い,内部に出血や壊死を認めることが多い。本腫瘍は髄膜皮細胞がある所であればどこでも発生しうるが,大脳半球円蓋部,傍矢状洞部,大脳鎌,小脳テント,鞍結節などが多い23)~29)。
3) 組織像および細胞像 ① 髄膜皮性髄膜腫(meningothelial meningioma)髄膜腫の基本的な組織像で,類円形核と淡好酸性の豊富な細胞質をもつ上皮様の細胞が大小の胞巣を形成しながら増殖する。胞巣内では渦巻き状配列(whorl formation)が目立つ。核は均一で多形性に欠き,細胞質は合胞状で境界不明瞭である(Figure 13.121)。圧挫標本においても,豊富な細胞質を有する上皮様細胞の大小のシート状集塊がみられる。集塊は重積性に欠き,集塊からほつれた孤在細胞もみられる(Figure 13.122)。これら孤在細胞では,淡い好酸性の多角形あるいは突起様の広い細胞質が明瞭に観察される。髄膜腫に特徴的な渦巻き状配列も目立ち,特異的ではないが核内細胞質封入体や核溝も観察される。核は類円形かつ均一で,細顆粒状の核網を呈し,核小体は目立たない(Figure 13.123)3),30),36),37)。

髄膜腫の基本的な組織型。各々の腫瘍細胞は合胞状で細胞境界が不明瞭で,胞巣状に増殖する。

上皮様集塊を形成する多角形の細胞質を有する腫瘍細胞。

特徴的な渦巻き状配列と核溝および核内細胞質封入体(矢印)。
線維芽細胞に類似した紡錘形細胞が膠原線維を伴いながら線維束を作り,平行配列や錯綜構造を示し増殖する。渦巻き状配列や上皮様細胞の胞巣状増殖は目立たない。膠原線維が豊富なため腫瘍組織は硬く,圧挫標本で薄く引き伸ばされないことが多い。シュワン細胞腫との鑑別が問題となる型(Figure 13.124)。

紡錘形細胞の束状配列や花むしろ状配列を認める。圧挫標本では束状集塊辺縁部での細胞のほつれがよく分かる。
最も頻度の高い組織型で髄膜皮性と線維性が混在したような組織像を示す。広い細胞質を有する腫瘍細胞が層板状に錯綜するため,組織標本では切れ方により束状配列ないしは胞巣状に見える。圧挫標本では合胞状の境界不明瞭な細胞質が豊富で,他の亜型に比べて渦巻き状配列が目立つ(Figure 13.125)。

合胞状の広い細胞質を有する細胞が層板状に錯綜あるいはシート状に増殖。渦巻き状配列が目立つ。
細胞質内に偽砂粒体(pseudopsammoma body)と呼ばれる好酸性の分泌物を有する腫瘍細胞が混在する髄膜腫。蝶形骨縁や前頭骨円蓋部などの頭蓋底に好発し,周囲脳に著明な浮腫を伴うことが多い。性差が顕著で7~8倍女性に多い。髄膜皮性あるいは移行性髄膜腫を基本構築とする組織および細胞像の中に,大小の分泌物を有する腫瘍細胞が混在する(Figure 13.126)。分泌物はPAS反応およびCEAに強陽性を示し,細胞質はcytokeratinに陽性でEMAに対する細胞膜の陽性強度も強い(Figure 13.127)23)~29),37)。

髄膜皮性あるいは移行性髄膜腫に類似した組織像の中に偽砂粒体と呼ばれる好酸性の細胞質内分泌物を有する上皮様細胞が混在する亜型。分泌物はPAS反応に陽性を示す。

細胞膜のリング状のEMA陽性所見は髄膜腫診断に有用な所見。分泌性髄膜腫の上皮様細胞はCEAに陽性を示す。
同心円状の石灰化物である砂粒体が大量に出現する髄膜腫。砂粒体の間は移行性髄膜腫に類似するが,細胞成分が少ない症例もある。細胞診標本にも多くの砂粒体が出現するため,圧挫標本を作製する際に硬く,ジャリジャリした感がある(Figure 13.128)。

移行性髄膜腫を基本とする組織構築の中に,極めて多数の砂粒体が認められる髄膜腫。
低異型度髄膜腫に増殖能亢進を示唆する退形成所見が加わった髄膜腫で再発率が高い。WHO分類では以下の診断基準が設けられている。1)核分裂像の増加(4個以上/高倍率10視野),2)組織学的な脳実質浸潤,3)次の5項目の中で3つ以上を満たす。①高い細胞密度,②N/C大の小型細胞,③明瞭な核小体,④構築に乏しいシート状増殖,⑤壊死巣(塞栓術に起因するものを除く)23)。圧挫標本での本亜型の診断は困難であるが,概して細胞結合性の低下を反映して,細胞集塊が小型で,孤在細胞の出現割合が高くなる。また多くの核分裂像や壊死が認められる場合は,本亜型の可能性が示唆される。
⑦ 脊索腫様髄膜腫(chordoid meningioma)脊索腫に類似した組織像を呈する髄膜腫。豊富な好塩基性の粘液様基質を背景に,空胞状の好酸性細胞質を有する腫瘍細胞が索状あるいは胞巣状に増殖する。圧挫標本では好塩基性およびGiemsa標本にて異染性を示す間質粘液が豊富で,小型の胞巣状および孤在性の腫瘍細胞を認める(Figure 13.129)。

粘液様の基質を背景に索状に配列する腫瘍細胞が増殖。腫瘍細胞は上皮様に配列し,空胞状の細胞質を有する。圧挫標本では,背景に異染性を示す間質粘液を認める。
ラブドイド細胞とは,横紋筋肉腫に類似した細胞で,腫瘍細胞は明瞭な核小体やクロマチン増加などの異型の強い偏在核を有し,細胞質は好酸性硝子様あるいは球状の封入体を持つ。封入体は中間径線維の塊でvimentinに強陽性を示す。これらの細胞が充実性に増殖し,多くの核分裂像や壊死を伴う23)~29),37)。急速に増大し,周囲脳への浸潤能も強く,遠隔転移も伴いやすい。予後不良で再発率が高く,約半数の症例が腫瘍死するといわれている(Figure 13.130)。

核異型が強い偏在核と封入体を伴う好酸性で厚みのある細胞質を有する。圧挫標本では集塊形成に乏しく,ほぼ孤在性に出現する。
血管周囲性の偽乳頭状配列が腫瘍の大部分を占める髄膜腫。血管から離れた細胞の虚血による壊死と離開によるため偽乳頭状増殖とされている。細胞密度が高く,核異型も強い。ラブドイド細胞が共存することもある。圧挫細胞診でも転移性腺癌との鑑別が必要となる程の核異型を示す細胞の乳頭状集塊が出現する。再発率が高く,遠隔転移をきたしやすい。小児・若年者に多いため,脈絡叢腫瘍や上衣腫との鑑別も重要である(Figure 13.131)。

血管周囲性の偽乳頭状配列が明瞭で,周辺に壊死巣を伴う。圧挫標本でも腫瘍細胞の血管周囲性の増殖が明瞭である。
脊索腫は骨腫瘍の一型で,胎生期脊索notochordの遺残組織から発生する悪性腫瘍である。遺残組織はトルコ鞍部から仙骨部まで分布しており正中線上の骨に好発する。頭蓋内発生は全脊索種の約40%を占め,その多くが斜体部に発生する。成人に多く,やや男性で多い。脊索腫は緩徐に増大する低悪性度肉腫であり,局所再発率が高く,肺や肝に遠隔転移することもある23)~29)。
2) 画像および肉眼所見(Figure 13.132)
T1で低信号を示す斜台部腫瘍。肉眼的に分葉状でゼリー様の粘液性の腫瘤で骨組織への破壊性に浸潤する。
MRI-T1強調で低信号,T2強調で高信号を示し,ガドリニウムで強く造影される。X線やCTでは骨破壊像がみられる。肉眼的には淡赤色のゼリー様の粘液性腫瘍で浸潤性増殖が強く,斜体部腫瘍では前方のトルコ鞍さらに蝶形骨洞まで浸潤し,後方では橋を圧排するように増殖する。
3) 組織像および免疫学的所見豊富な粘液性基質の間質を背景に,結合性を有する上皮様細胞が索状,コード状および胞巣状に増殖する(Figure 13.133)。腫瘍細胞の細胞質は淡好酸性で空胞状を示し,グリコーゲンおよび中性ムチンを多く含みPAS反応陽性である。細胞質内に多数の空胞を有する細胞は担空胞細胞(physaliphorous cell)と呼ばれ,本腫瘍における診断根拠として意義が高い。間質の粘液はアルシアン青およびトルイジン青陽性を示す。免疫化学的に腫瘍細胞は,S100蛋白,EMA,CEA,cytokeratin(CK8, CK18, CK19)に陽性で,脊索への分化を司る転写因子で,特異度の高いマーカーであるbrachyuryが核に陽性を示す(Figure 13.134)。

粘液様基質の間質の中に結合性が強く,豊富な淡好酸性の細胞質を有する上皮様の腫瘍細胞が索状・胞巣状に増殖。

間質粘液はA.blue陽性。腫瘍細胞はPAS,brachyuryに陽性。
時に豊富な軟骨性基質を伴うことがあり,軟骨様脊索腫(chondroid chordoma)と呼ばれる。本型は通常の脊索腫と比べて予後が良いとされているが,若年者で多く発生し,予後に差はないという報告もある(Figure 13.135)。低分化型脊索腫(poorly differentiated chordoma)および未分化肉腫成分を併せ持つ脱分化型脊索種(dedifferentiated chordoma)では,遠隔転移や局所再発率が高く予後不良である。一部の低分化型脊索腫で22番染色体長腕のSMARCB1/INI1遺伝子の欠失がみられることがあり,FISH法あるいは免疫染色でのINI1蛋白の核の陰性化より証明される26)。

背景に豊富な軟骨基質を認める軟骨様脊索腫。
鑑別診断として,特に軟骨様脊索腫では軟骨肉腫が挙げられるが,軟骨肉腫では細胞の結合性に乏しく,上皮様集塊がみられず,EMA,cytokeratinなどの上皮マーカーの発現はみられない。また非常に稀ではあるが,脊索腫様髄膜腫や腎癌および粘液産生性の腺癌との鑑別も重要である。いずれの症例もbrachyuryに陰性を示すため本マーカーの免疫学的検索は不可欠である。
斜体部には,胞状外脊索症(ecchordosis physaliphora)と呼ばれる先天性の過誤腫性の微小嚢胞性病変が認められることがあるが,この組織像は脊索種に極めて類似しており両者の鑑別は困難である。剖検時に偶発的に発見されることがほとんどであるが,その頻度は健常人の1~2%と決して低くない。本病変の脊索腫との鑑別点は増大することがなく,骨組織への浸潤像がない点である28),29)。
4) 細胞像捺印および圧挫標本では,ヘマトキシリンに淡染する粘液様物質を背景に豊富な細胞質を有する大型の上皮様細胞が散在性にあるいは弱い結合性を示す小集塊として出現する(Figure 13.136)。集塊は重積性に欠くシート状あるいは索状に配列し,細胞境界は明瞭な細胞と不明瞭な細胞が混在する。細胞質は淡明あるいは淡い好酸性を示し,PAS反応陽性で多くのグリコーゲン顆粒を認める(Figure 13.137)。核は類円形で異型性に乏しいのが一般的であるが,症例によっては核の大小不同,明瞭な核小体,粗大顆粒状クロマチンの増量,核膜の肥厚,多核細胞を認める(Figure 13.138)。細胞質内に多数の大小の空胞を有する担空胞細胞は,脂肪芽細胞に類似し,細胞境界は比較的明瞭である(Figure 13.139)。

粘液性の背景に淡明な腫瘍細胞が上皮様に集簇して出現。
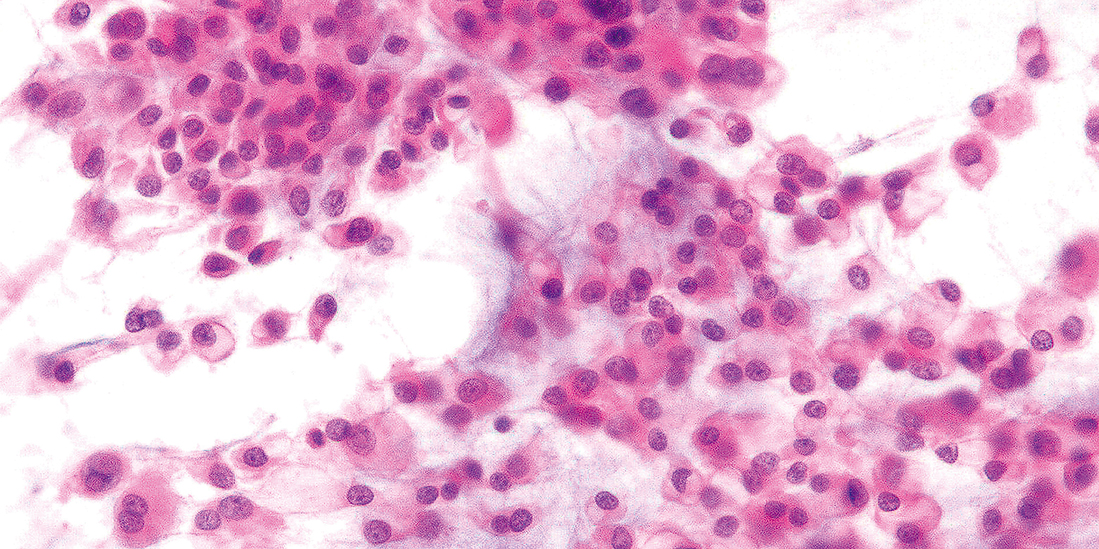
個々の腫瘍細胞は互いに結合性を示し,シート状の集塊を形成する。細胞質は豊富で好酸性を示す。

症例によっては,核の大小不同,明瞭な核小体,多核細胞,粗顆粒状クロマチンの増量を認める。

細胞質はグリコーゲンを多く含むため淡明で辺縁不明瞭なレース状を示す。大小の空胞を有する細胞は担空胞細胞と呼ばれ,本腫瘍診断の重要な鍵となり得る。
鑑別診断としては,組織診断と同じく軟骨肉腫や転移性腺癌が挙げられるが,軟骨肉腫の方がより細胞が小型かつ均一で,上皮様結合に乏しく,細胞質内の空胞も目立たない。また転移性腺癌では核異型ならびに多形性が強く,重積性を示す大型集塊として出現し,核分裂像や壊死の出現程度が高い3),38)。
13. 原発性中枢神経系リンパ腫(primary central nervous system lymphoma; PCNSL) 1) 概要PCNSLは中枢神経系に発生する節外性リンパ腫の一つであり,二次的に頭蓋内に進展してきたものは除外される。90%以上が中枢神経系びまん性大細胞型B細胞リンパ腫(DLBCL of the CNS)で,稀な型としてはHIVあるいはEBV感染に関連した免疫不全症随伴性中枢神経系リンパ腫,血管内大細胞型B細胞リンパ腫などがある。DLBCL of the CNSは50~70歳の高齢者に多く,5年生存率は20%程度で予後不良である。近年,発生頻度が増加している23)~27)。
2) 画像および肉眼所見(Figure 13.140)
造影MRI(左)で強く増強され,PET-CT(右)でも高度に集積する境界明瞭な腫瘤。周囲には浮腫を伴っている。
MRI-T1強調で低信号,T2強調で等~高信号を示し,造影で強く増強される。PET-CTでも著しく高い集積を認める。大脳半球に好発し,しばしば多発性の病巣を形成する。
3) 組織像および免疫学的所見(Figure 13.141)
血管周囲に高密度に集簇し,周囲に壊死巣を伴う。腫瘍細胞は円形で,粗顆粒状クロマチンが増量し,核小体が明瞭。
極めて細胞密度の高い腫瘤を形成する。血管周囲のVirchow-Robin腔に腫瘍細胞が集簇して増殖するいわゆるperivascular cuffingが特徴的である。種々の程度で地図状壊死巣を認める。脳実質内にびまん性に浸潤し,周囲には星細胞増生を伴う。腫瘍細胞は通常のDLBCLと同じく大型類円形でN/Cが高く,明瞭な核小体,粗大顆粒状クロマチンの増量,核の切れ込み像が目立ち,核分裂像やアポトーシス像を多数認める。免疫化学的にCD20,CD79aなどのB細胞マーカーに陽性で,Bcl-2,Bcl-6,c-Mycが高発現する増殖活性の高い形質を示すことが多い23)~27)。
4) 捺印細胞像(Figure 13.142, 13.143)
リンパ腫細胞に加えて,組織球や星細胞の増生もみられる。

M-G標本では著明な核形不整,打ち抜き空胞像が目立つ。
圧挫標本より捺印標本の方が観察しやすい。細胞像は他臓器におけるDLBCLと同様で,Giemsa標本での好塩基性の強い細胞質と核および細胞質の打ち抜き空胞像が目立つ。壊死巣や腫瘍辺縁部では多数の泡沫組織球や反応性星細胞の増生を伴うため,脱髄性疾患や感染症との鑑別が問題となることがある。核異型の強いリンパ球を認識できれば診断は容易である。また一部の膠芽腫や小細胞癌の転移との鑑別がしばしば問題となるが,圧挫標本における細胞質突起および背景のglio-fibrillary backgroundの有無,細胞接着性や集塊形成の評価が重要である。
14. 胚腫(ジャーミノーマ)(germinoma) 1) 概要生殖腺や縦隔に発生する胚細胞性腫瘍同様に原始胚細胞に由来する腫瘍で,中枢神経系胚細胞腫瘍central nervous system germ cell tumor(CNS GCT)と総称される。脳の正中部である松果体,視床下部,トルコ鞍上部に好発し,20歳未満の小児・若年者に多い。組織型としては奇形種,胚腫(ジャーミノーマ),卵黄嚢腫瘍,胎児性癌,絨毛癌があり,複数のGCTが混在する腫瘍は混合型胚細胞性腫瘍とされ,予後不良である。血清の腫瘍マーカーとして卵黄嚢腫瘍ではα-fetoprotein,絨毛癌ではβHCGが高値を示す。純粋型の胚腫は放射線や化学療法の感受性が高く予後は比較的良好である(10年生存率は約80%)23)~29)。本項では頻度の高い胚腫について概説する。
2) 画像および肉眼所見(Figure 13.144)
松果体部に強く均一に増強される腫瘤を認める。
MRI-T1強調で低~等信号,T2強調で高信号を呈し,均一に強い造影効果を示す。肉眼的には灰褐色調の充実性腫瘤で出血を伴うことが多い。
3) 組織像および免疫学的所見(Figure 13.145)
リンパ球浸潤が強い二相性(左)がみられる。大型で淡明な細胞質を有する腫瘍細胞がシート状~敷石状に増殖する。
大型核と淡明な細胞質を有する腫瘍細胞が敷石状あるいはシート状に増殖する大小の腫瘍胞巣を形成する。間質には小型成熟リンパ球の浸潤が著しい,いわゆるtwo cell patternを示す。核クロマチンは極めて繊細で,大型の核小体を有し,核膜は薄い。明るい細胞質にはグリコーゲンが豊富でPAS反応で顆粒状の陽性を示す。組織球や類上皮細胞の増生を伴った肉芽腫性変化が強い症例もある。約10%の症例で合胞体性栄養膜細胞syncytiotrophoblastic giant cell(STGC)がみられる。免疫染色にて腫瘍細胞はplacental alkaline phosphatase(PLAP),c-kit(CD117),podoplaninに細胞膜が陽性を示し,Oct4に核陽性を示す(Figure 13.146)。STGCはβHCGに陽性である。

細胞膜がPLAPに陽性で,核にOct4が発現している。

腫瘍細胞は淡明で豊富な細胞質を有し,非常に繊細・緻密なクロマチンが増量し,著しく腫大した核小体が明瞭である。
多くの小型成熟リンパ球と共に,大型異型細胞が弱い結合性を示す敷石状集塊を形成あるいは孤在性に出現する。類円形~多角形の広い淡明な細胞質を有し,細胞膜が明瞭で個々の細胞境界も明確。核形不整に乏しく,非常に繊細緻密な核クロマチン網が悪性リンパ腫を含めた他の悪性腫瘍と異なる3),38)。
15. 頭蓋咽頭腫(craniopharingioma) 1) 概要胎生期のラトケ嚢上皮遺残から発生すると考えられているトルコ鞍上部に好発する上皮性の嚢胞性腫瘍でエナメル上皮腫型(adamantinomatous type)と乳頭型(papillary type)の二型があり,いずれも良性腫瘍である(WHO grade I)。頭蓋内腫瘍の約2~3%の頻度で,このうちの約1/4の症例が15歳未満の小児でみられ,小児原発性脳腫瘍の約10%を占める。エナメル上皮腫型は小児と成人に二峰性の分布を示す(小児では5~10歳にピーク,成人では50歳台にピーク)。乳頭型は成人に多い傾向がある23)~29)。
2) 画像および肉眼所見エナメル上皮腫型はCTで石灰化が目立つ充実部を含む多房性の嚢胞性腫瘤を形成することが多く,MRIでは充実部および嚢胞壁が強く増強される。肉眼的に充実部は分葉傾向を示し,嚢胞内容液は粘度のある黄~緑褐色調の液体で「モーターオイル様」と形容される。充実部分は石灰化や白色調の角化物を含む(Figure 13.148)。乳頭型は境界明瞭な充実性腫瘤で,石灰化や嚢胞形成に乏しい。

一部に強く造影される多房性の嚢胞性腫瘤を形成する(左)。
充実部分には石灰化や白色の角化物の沈着を伴うことが多い(右)。
エナメル上皮腫型頭蓋咽頭腫(Figure 13.149)では,以下の3種類の特徴を示す細胞がみられる。一つは胞巣の最外層で柵状に配列する高円柱状の細胞で,二つ目はその内部に胞巣状,渦巻き状に配列する重層扁平上皮の有棘細胞に類似した細胞で,これらは細胞間に空陵が目立ち,細胞同士が網目状に連結して星芒状に見えるためstellate reticulumと呼ばれる。三つ目が染色性を失った核と好酸性細胞質を有する陰影状上皮細胞の角化物の塊でwet keratinと呼ばれ,本腫瘍に極めて特徴的かつ診断特異的な所見である。上皮細胞巣およびwet keratin内にはしばしば石灰化を伴う。疎な結合組織よりなる間質にはリンパ球,泡沫組織球および多核巨細胞の浸潤,コレステリン結晶の沈着を認めることが多い。また腫瘍と接する周囲の脳実質内にはRosenthal fiberを伴った反応性星細胞増生を認め,piloid gliosisと呼ばれる。

胞巣の辺縁部では柵状に並ぶ円柱状細胞,内部では星芒状の細胞が網目状に増殖する。好酸性角化物の塊であるwet keratinが特徴的。
乳頭型頭蓋咽頭腫(Figure 13.150)では非角化型重層扁平上皮細胞が大小の線維性血管間質を伴って乳頭状に増殖する。エナメル上皮型でみられる円柱状細胞の柵状配列,stellate reticulumやwet keratinは認められない。個々の扁平上皮細胞では細胞間橋が明瞭で核異型に乏しく均一な所見である23)~29)。

血管結合組織を取り囲む扁平上皮細胞の乳頭状増殖。
上皮細胞はEMA,cytokeratin(CK5/6,CK7,CK14)に陽性を示す。エナメル上皮腫型ではβ-cateninの発現の局在が細胞膜のみならず核での発現を認め,この所見は特に渦巻き状配列を示す内部の星芒状細胞でみられる(Figure 13.151)。一方乳頭型では細胞膜にのみリング状の陽性所見を示す(Figure 13.152)。

特に渦巻状集塊部の細胞でβ-cateninの核局在がみられる。

乳頭型では,細胞膜にのみβ-cateninの発現がみられる。
エナメル上皮腫型の95%にもおよぶ症例でCTNNBI遺伝子(β-cateninをコード)の3番目のexonに変異があることが判明しており,これに伴うWNTシグナル伝達系の活性化が腫瘍発生に関与しているとされている。また乳頭型ではBRAF遺伝子V600E変異が約90%以上の症例で認められる。この2つの遺伝子異常は相互排他的で各々の亜型に特異的な所見である23),26),27)。
6) 圧挫細胞像エナメル上皮腫型(Figure 13.153)では嚢胞性病変を示唆するリンパ球や泡沫組織球を含む粘液様物質を背景に大小の集塊を形成する腫瘍細胞を認める。集塊は扁平上皮細胞に類似した重厚な細胞質を有する紡錘形~多角形の細胞と集塊辺縁部で柵状配列を示す円柱状細胞より構成される。さらにHE染色で淡赤色~黄褐色調,Pap染色で黄緑色~橙色を呈し,高輝度の顆粒状を示すwet keratinを認める。

紡錘形~多角形の細胞(左)と集塊辺縁部での柵状配列が明瞭な円柱状細胞(右)およびwet keratinがみられる。
乳頭型(Figure 13.154)では孤立散在性あるいは大小のシート状および乳頭状集塊を形成する扁平上皮細胞を認める。重厚かつ好酸性の角化した多角形の細胞質には小型で異型に乏しい核を認める。個々の細胞境界は明瞭で,集塊内での重積性は乏しい。

多角形の広い細胞質を有する扁平上皮細胞のシート状増殖がみられる。核は小型でN/Cが低く,異型性に乏しい。
鑑別診断として扁平上皮細胞が出現するラトケ裂嚢胞(Rathke’s cleft cyst),類表皮嚢腫(epidermoid cyst)や皮様嚢腫(dermoid cyst)が挙げられるが,ラトケ裂嚢胞では線毛あるいは粘液を有する高円柱状細胞が孤在性に認められ(Figure 13.155),また類表皮嚢種や皮様嚢種での扁平上皮細胞にはケラトヒアリン顆粒を認めることが多い。いずれの病変においてもwet keratinがみられない点が重要な所見である3),38)。

エナメル上皮腫型頭蓋咽頭腫(左)では,緑色~黄緑色調で輝度の高い顆粒状を呈する大小のwet keratinがみられるが,ラトケ裂嚢胞(右)では認められず,線毛円柱上皮や粘液含有細胞がみられる。
下垂体前葉のホルモン産生細胞から発生する良性腫瘍で,原発性脳腫瘍の約20%を占める頻度の高い腫瘍。臨床的にホルモン分泌過剰による血中濃度の上昇を伴う機能性腺腫と分泌過剰を示さない非機能性腺腫とに分けられる。病理学的には産生するホルモンによって主に以下のように分類される23)~27)。
・成長ホルモン細胞腺腫(somatotroph adenoma; GH)
・プロラクチン細胞腺腫(lactotroph adenoma; PRL)
・甲状腺刺激ホルモン細胞腺腫(thyrotroph adenoma; TSH)
・副腎皮質刺激ホルモン細胞腺腫(corcicotroph adenoma; ACTH)
・性腺刺激ホルモン細胞腺腫(gonadotroph adenoma; LH, FSH)
・ホルモン陰性腺腫(null cell adenoma)
2) 画像および臨床所見(Figure 13.156)
MRI-T1強調で高信号を示す腫瘍。トルコ鞍の拡大を認める(左)。
単純X線で足底部軟部組織厚(heel pad)の増大がみられる(右)。
MRI-T1強調で低~高信号,T2強調で高信号を示す。腫瘍径が10 mm以上のmacroadenomaではトルコ鞍の骨破壊,髄膜や海綿静脈洞への浸潤がみられる。巨人症や先端肥大症といった特徴的な臨床像を示す成長ホルモン細胞腺腫では,単純X線でのトルコ鞍の拡大および破壊,副鼻腔の拡大,下顎角の開大や足底部軟部組織厚heel padの増大(22 mm以上),外観的には鼻・口唇の肥大,巨大舌,下顎の突出などが認められる。プロラクチン細胞腺腫では,乳汁過多や無月経,副腎皮質刺激ホルモン細胞腺腫ではCushing症候群やNelson症候群を呈する。
3) 下垂体腺腫の基本的な組織像正常の下垂体前葉では,数十個の細胞で構成される小葉あるいは腺房状の構築がみられるが,腺腫ではこれらの構造が破壊され,びまん性,血管周囲性(偽乳頭状),柵状配列などの構造を示す。びまん性は機能性腺腫,特にGH細胞性に多く,血管周囲性は非機能性やACTH細胞性で多くみられる(Figure 13.157)。また細胞質の染色性からエオジンやオレンジGに好染する好酸性,PAS反応陽性を示す好塩基性,HEで淡桃色を示す嫌色素性に分類され,好酸性はdensely granulated typeのGH細胞性に多く(Figure 13.158),好塩基性はdensely granulated typeのACTH細胞性で多くみられる。核はいずれも異型に乏しく,類円形で均一であるが,多形性に富む症例もある。一部のTSH細胞性や非機能性腺腫において紡錘形の形態が目立つ症例もある。

びまん性(左),敷石状,柵状配列,血管周囲性の偽乳頭状(右)など様々な増殖形態を示す。各々の細胞形態は極めて単調である。

HEでエオジン好性を示す腫瘍細胞がオレンジ色に染色される。
免疫化学染色は本腫瘍の亜型分類に必須である。GH細胞腺腫ではGHに加えて,cytokeratinに陽性を示す中間径細線維の凝集物でfibrous bodyと呼ばれる細胞質内の封入体様の構造物を認める(Figure 13.159)。これはGH細胞腺腫の中でも好酸性顆粒状の分泌物に乏しいsparsely granulated typeで多く認められる。また近年では下垂体前葉幹細胞から各々のホルモン産生細胞へ分化する転写因子も明確になっており,GH,PRL,TSH細胞性ではPit-1,ACTH細胞性ではTpit,LH,FSH細胞性ではSF-1がそれぞれ核に陽性を示す23)~26)。

細胞質はGHに顆粒状に陽性。封入体構造はCAM5.2陽性を示す。
比較的豊富な細胞質を有する類円形細胞の均一な増殖を認める。核クロマチンは繊細な顆粒状で小型の核小体を認める。細胞質は顆粒状で厚みがあり境界明瞭なもの,繊細で境界不明瞭なものがある。
GH細胞腺腫では特徴的な細胞所見を呈する。細胞質は好酸性で顆粒状を示し,核は偏在し,2核細胞が目立つ。他の腺腫に比べてやや細胞の大小不同が目立つ傾向にある(Figure 13.160)。細胞診標本でも核近傍の好酸性の封入体様構造(fibrous body)が明瞭に観察され,また相互封入像も目立つ(Figure 13.161)。

好酸性の細胞質を有する類円形腫瘍細胞の単調な増殖よりなる。

核は偏在し,好酸性の細胞質はやや顆粒状を示す。核周囲に類円形の封入体構造(fibrous body)を認める。GH細胞腺腫では二核細胞,相互封入像(右矢印)が他の腺腫と比べ数多く出現する特徴がある。
PRL細胞腺腫では特徴的な所見には乏しく,その他の腺腫との形態学的な鑑別は困難であるが,細胞質がより淡く,境界不明瞭で,やや多角形~紡錘形の細胞質を示すことが多い(Figure 13.162)。

細胞質は淡く,繊細で境界不明瞭。やや紡錘形を示す細胞も混在。
ACTH細胞腺腫では,血管周囲性の偽乳頭状配列を示すことが多い組織像を反映して,圧挫標本では血管周囲に腫瘍細胞が集簇する傾向が強い。ACTH細胞腺腫の多くは腫瘍径10 mm以下のmicroadenomaのことが多く,densely granulated typeで細胞質は好塩基性でPAS反応陽性を示すことが多い。非腫瘍組織のACTH細胞は萎縮し,高コルチゾール血症に起因するネガティブフィードバック機構によりしばしば細胞質内にcytokeratin filamentが集積し,この現象はCrooke変性と呼ばれる,ACTH細胞性macroadenomaの中で多くの細胞にCrooke変性が認められる亜型が存在し,Crooke cell adenomaと呼ばれる。この腺腫は増大傾向が強く浸潤性で再発しやすく,中年女性に多いといわれている。組織・細胞診標本ともに硝子様で重厚な細胞質が特徴的である(Figure 13.163)39)。

ACTH細胞腺腫の一亜型。細胞質は硝子様で厚みのある淡い好酸性を示しcytokeratinにリング状の陽性を示す。分泌顆粒は極僅かで,細胞質中心部あるいは辺縁部に集簇する傾向が強い。
本論文に関連し,開示すべきCOI 状態にある企業等はありません。
松本 慎二(福岡大学病院)…I, II, IV
鍋島 一樹(福岡大学病院)…I, II, IV
井上 亨(福岡大学病院)…I, II, IV
冨永 晋(防衛医科大学校)…III
担当編集委員青木 裕志(順天堂大学)