Abstract
The nature of Si-O bonding and Si-O-Si bridging is discussed using molecular orbital
calculations. We found the equilibrium geometries for two pyrosilisic acid molecules
(C2V and 60° torsion) using Møller-Plesset perturbation theory and 6-311G
(d,p) split valence basis set. The bent configuration of the Si-O-Si angle in equilibrium
geometries can be explained by the balance of Coulombic repulsion between SiO4
tetrahedra and the energy of lone pair orbitals belonging to bridging oxygen atom without
concerning the contribution of d-p π-bonding from the results of natural bonding orbital
analysis. The energy surfaces of two pyrosilisic acid molecules with varying Si-O length
to the bridging oxygen and Si-O-Si angle were calculated and we found the relationship
between Si-O length to the bridging and Si-O-Si bridging angle. The Si-O bonding
strengthens with increasing Si-O-Si angle because of stabilization in energy of Si-O
bonding orbital with decreasing the hybridization index λ in spλ orbital of
bridging oxygen and increase of Coulombic interaction between Si and bridging oxygen
atom.
1 Introduction
Understanding the nature of Si-O bond and Si-O-Si bridging is important for mineralogy,
material science and metallurgy. It is well known that the variation of Si-O-Si angle in
silicates is caused by difference of composition, temperature and pressure. The change in
angle of Si-O-Si bridging affects the strength of Si-O bond. For instance, decrease of
Si-O-Si angle increases the Si-O bond length in coesite crystal (Gibbs et
al. [1]). The decrease of Si-O-Si angle of
liquid silicates as a result of compression was reported by various researchers
(e.g., Navrotsky et al. [2], Ohtani et al. [3],
Sakamaki et al. [4]). The decrease
of Si-O-Si angle is thought to be the trigger of decrease of viscosity of liquid silicates
(Navrotsky et al. [2],
Noritake et al. [5]). Quantum
chemical properties of Si-O-Si bridging was investigated to understand the relationship
between Si-O-Si angle, Si-O bond length and its strength (e.g., Newton and
Gibbs [6], Tsuneyuki [7], Kubicki and Sykes [8]). Newton and Gibbs
[6] reported that a pyrosilisic acid molecule has
energy minimum at Si-O-Si angle of 145° using Hartree-Fock method and STO-3G basis set
(Hehre et al. [9]). Tsuneyuki
[7] reported that the bent configuration of Si-O-Si
angle in pyrosilisic acid molecule is not reproduced using double-zeta function basis set
nevertheless increase of the number of basis function generally increases reproducibility.
The relationship between Si-O bond length and Si-O-Si angle, that is, the nature of Si-O-Si
bridgings seems not to be reproduced by increase of basis function using Hartree-Fock
method. In this paper, we show the molecular orbital calculations about the pyrosilisic acid
molecule using post-Hartree-Fock method and more precise basis set to understand the nature
of Si-O-Si bridging.
2 Method
Molecular orbital calculations were performed using the Gaussian 09 code [10]. We first calculate the optimized structure of
disiloxane molecule by Hartree-Fock (HF), second-order Møller-Plesset perturbation theory
(MP2) [11], and two density functional theory
(three-parameter hybrid functional (B3LYP) [12] which
is a mixture of Hartree-Fock exchange with DFT exchange functional by Becke and correlation
functional by Lee, Yang and Parr [13], and
generalized gradient approximation by Perdew, Burke and Ernzerhof (PBE) [14]) with 6-311G (d,p) split valence basis set [15,16]. The values
of structural parameters of disiloxane molecule optimized by various models and experimental
data by Almenningen et al. [17] are
shown in Table 1. The Si-O-Si angle is not
reproduced by HF method as shown in Tsuneyuki [7]. The
bent configuration of Si-O-Si angle is reproduced by use of MP2 and density functional
theory with PBE. The optimized angle of Si-O-Si in disiloxane molecule by MP2 is closer to
the experimental value than that by PBE. Then we apply the MP2 method with 6-311G (d,p)
basis set to the calculation of the pyrosilisic acid molecule,
H6Si2O7. Kubicki and Sykes [8] reported that hydrogen bonding is made in the molecule in optimizing
the molecular structure of pyrosilisic acid. We optimized pyrosilisic acid molecule with
z-matrix to define the point symmetry of pyrosilisic acid molecule to avoid the effect of
hydrogen bonding. Two types of structure were optimized, one is the C2v point
symmetry as shown in Figure 1a, the other is
structure in which each tetrahedra is in conformational relation of 60° torsion as shown in
Figure 1b. The optimized structural parameters
are shown in Table 2.
Table 1.
The optimized parameters and difference from experimental value in angstrom and
degree of disiloxane molecule, (SiH
3)
2O by MO calculation using
various model chemistry. The experimental values are taken from Almenningen
et
al. [
17].
|
Exp. |
HF |
Δ |
MP2 |
Δ |
B3LYP |
Δ |
PBE |
Δ |
| Si-O |
1.634 |
1.621 |
−0.013 |
1.642 |
0.008 |
1.640 |
0.006 |
1.635 |
0.001 |
| Si-H |
1.486 |
1.476 |
−0.010 |
1.476 |
−0.010 |
1.485 |
−0.001 |
1.488 |
0.002 |
| Si-O-Si |
144.1 |
180.0 |
35.9 |
156.5 |
12.0 |
179.2 |
35.1 |
169.7 |
25.6 |
| O-Si-H |
109.9 |
109.9 |
0.0 |
110.4 |
0.5 |
109.9 |
0.0 |
110.1 |
0.2 |
| H-Si-H |
109.1 |
109.0 |
−0.1 |
109.0 |
−0.1 |
109.0 |
−0.1 |
109.0 |
−0.1 |
Table 2.
The optimized parameters in angstrom and degree of pyrosilisicacid molecule,
H
6Si
2O
7 by MO calculation using MP2 level and
6-311G(d,p) basis set.
|
C2V |
60° torsion |
| Si-Obr |
1.601171 |
1.604154 |
| Si-Onbr |
1.648782 |
1.648563 |
| O-H |
0.954669 |
0.954561 |
| Si-O-Si |
172.006 |
159.647 |
| Si-O-H |
117.705 |
117.857 |
| Total Energy (Hartree) |
−1107.5476706 |
−1107.5482842 |
Then we calculate the potential energy surfaces of two pyrosilisic acid molecules varying
the Si-Obr bond length and Si-O-Si angle with fixing other structural parameters
obtained by optimization. To analyze the nature of bonding, we use the Natural Bonding
Orbital (NBO) analysis method (Foster and Weinhold [18], Reed et al. [19,
20]). NBO analysis originated as a technique for
studying hybridization and covalency effects in molecular wave functions. In NBO analysis,
the calculated molecular orbitals are decomposed and recomposed visualized bond orbital that
corresponds to the picture of interatomic bonds and lone pairs. It gives us energy of
bonding, degree of hybridization of bonding, overwrap integration weighted bond order and
charge of atoms. In NBO analysis, we firstry define natural atomic orbital and attribute the
electrons. Natural atomic orbital can be obtained by block diagonalization of density matrix
by each angular momentum of atomic orbital of each atom. Then we define the spλ
natural hybrid orbitals, hλ(θ) as follows,
|
h
λ
(
θ
)
=
N
(
s
+
λ
1
2
p
θ
)
| (1) |
where
pθ is a
normalized
p orbital pointing in the direction
θ and N is
a normalization constant. Then we can obtain the natural bond orbital
σAB between atom
A and
B
from directed orthonormal hybrid orbitals
hA and
hB (natural hybrid orbitals) as follows,
|
σ
A
B
=
c
A
h
A
+
c
B
h
B
| (2) |
where c is the coefficient calculated
numerically from the result of density by molecular orbital calculations. More precise
information about NBO analysis is written in [
18,
19,
20].
3 Results and Discussion
The optimized structural parameter of the pyrosilisic acid molecules is shown in Table 2. Stable Si-O-Si angle and total energy of
C2V model are larger than those of 60° torsion model. However, the bond length
between Si and bridging oxygen (Obr) is not different significantly by the
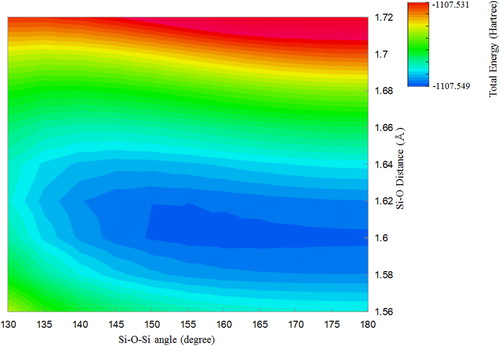
torsion angle. Figures 2 and 3 are contour maps of
the potential energy surface of the pyrosilisic acid molecules (C2V and 60°
torsion models). The stable Si-O bond lengths decrease with increasing Si-O-Si angle in both
molecules. Figure 4 shows the sum of orbital
energy of valence electrons belonging to bridging oxygen, energy of each valence orbital,
and overlap weighted bond order of Si-Obr. The sum of orbital energy of valence
electrons belonging to the bridging oxygen decreases with decreasing Si-O-Si angle.
Decomposing into each orbital, orbital energy of sum of two lone pairs decreases with
decreasing Si-O-Si angle, however the energy of Si-O bonding orbital increases with
decreasing Si-O-Si angle. Note that the calculated orbitals are decomposed based on natural
Lewis structure and lone pairs are orthogonal with each other. The overlap weighted bond
order has local minimum at 140 to 145 degrees whereas the changes in orbital energies change
monotonously. Calculated charges of Si and Obr and electrostatic potential
between Si and bridging oxygen calculated using charges are shown in Figure 5. In NBO analysis, the charge at each atom is calculated from
occupancy of natural atomic orbital. The absolute value of atomic charge increases with
increasing Si-O-Si angle. The order of changes in electrostatic potential between Si and
bridging oxygen with varying Si-O-Si angle is comparable to that in the energy of Si-O
bonding orbital. Figure 6 shows the coefficient
cSi and cO in equation (2) with varying Si-O-Si angle. The
coefficient of cSi decreases and cO increases with increasing Si-O-Si
angle.




The bent configuration of Si-O-Si bridging is reproduced in this study using Møller-Plesset
perturbation theory and 6-311G (d,p) split valence basis set. The bent configuration of
Si-O-Si bridging is often explained by three contributions, Coulombic repulsion between two
SiO4 tetrahedra, lone pair (or valence shell electron pair repulsion rule) of
bridging oxygen, and the d-p π-bonding model. The contribution of Coulombic repulsion to
energy surface seems to be important. Because the bent configuration cannot be reproduced in
some combinations of model chemistry and basis set (Tsuneyuki [7]). The contribution of lone pair electrons of bridging oxygen decreases
the bridging angle. The oxygen atomic orbitals make sp3 hybrid orbitals that form
tetrahedral configuration constructed by two bonding orbitals and two orbitals with lone
pairs because of Coulombic repulsion between these orbitals. This stabilization with
decreasing Si-O-Si angle is represented in Figure
4. The sum of the energy of valence orbitals decreases with decreasing Si-O-Si
angle. The energy of lone pair orbitals split with decreasing Si-O-Si angle, but one of them
does not change because of the orthogonal decomposing of lone-pair electrons by NBO method,
unlike with bonding orbitals. However the changes in orbital energy of valence orbital of
oxygen atom shows the tendency of equalization of each orbital. Consequently the effect of
lone pairs potentially narrows the Si-O-Si bridging angle. The decrease in the energy of
lone pair orbital is comparable to the electrostatic potential of the molecule calculated
from charge of atom by NBO analysis (Figure 7).
The sum of the electrostatic potential calculated from charge and the energy of lone pair
electron has local minimum around 165 degrees. The d-p π-bonding between Si and O atoms is
considered to play an important role in Si-O-Si bridging [21]. Newton and Gibbs [6] calculated the
contributes of 3d-orbital to Si-O bond by Mulliken population analysis [22] from the results of MO calculation with Hartree-Fock
method and STO-3G basis. In Newton and Gibbs [6], the
total overlap populations in Si-O bond is 0.832 to 0.862. The population of σ-bonding
between sp hybrid orbital of Si and O is 0.556. The populations of σ-bonding and π-bonding
which 3d-orbital involves are 0.113 and 0.163, respectively. The occupancy of valence
orbitals analyzed by natural atomic orbital analysis is shown in Table 3. The ratio of occupancy of d-orbital to s- and p-orbital
is about 1% whereas the population analysis by Newton and Gibbs [6] shows several tens of percent. The effect of d-p π-bonding seems to be
negligible in Si-O-Si bridging. Consequently, the bending of Si-O-Si bridging can be
explained by the balance of Coulombic repulsion between SiO4 tetrahedra and lone
pair electrons of bridging oxygen atom.

Table 3.
The occupancy of natural atomic orbital of Si in various Si-O-Si angle in
pyrosilisic acid molecule (60° torsion model).
|
130° |
155° |
180° |
| 3s |
0.44388 |
0.43775 |
0.43546 |
| 3px + 3py + 3pz |
0.92376 |
0.91504 |
0.91303 |
| 3dxy + 3dyz + 3dzx + 3dx2y2 +
3d2 |
0.05959 |
0.05834 |
0.05808 |
The general result of works about the nature of Si-O-Si bridging is Si-O bond lengthens
with decreasing Si-O-Si angle [2,6,23]. This phenomenon is also
reported by experiments [1]. Our study shows the same
tendency in energy surface map (Figure 2 and 3).
The mechanism of Si-O bond lengthening with decreasing of Si-O-Si angle can be explained by
the energy of Si-O bonding orbital, overlap weighted bond order and Coulombic
interaction.
The energy of Si-O bonding orbital increases with decreasing Si-O-Si angle (Figure 4). This means that the Si-O bonding stabilizes
when the Si-O-Si angle is 180°. The ideal energy of sp hybrid orbital should be lower than
that of sp3 hybrid orbital because the energy of s-orbital is lower than that of
p-orbital with the same principal quantum number, qualitatively. The changes in orbital
energy during decrease of Si-O-Si angle might be the result of increase of ratio of p
character in spλ hybrid orbital in bridging oxygen. The hybridization may be
considered to make Si-O bond weakened with decreasing Si-O-Si angle, from the viewpoint of
energy of bonding orbital.
Another view point is overlap-integration weighted bond order. An increase of overlap
weighted bond order increases bond strength especially in the case of homonuclear diatomic
molecule. The calculated overlap weighted bond order of Si-Obr bonding has local
minimum at Si-O-Si angle of 140 to 145 degrees whereas the energy of Si-O bonding orbital
monotonously increases with decreasing Si-O-Si angle (Figure 4). However the decrease of overlap weighted bond order can weaken the Si-O
bonding at least at high-angle region.
The changes in the charge of atom (Figure. 5) seem to support the weakening of Si-O bonding
with decreasing Si-O-Si angle because the absolute value of charge of Si and bridging oxygen
decreases with decreasing Si-O-Si angle.
Consequently the Si-O bond weakens and lengthens with decreasing Si-O-Si angle because of
stabilization of Si-O bonding orbital with decreasing the hybridization index λ in
spλ orbital of bridging oxygen and increase of Coulombic interaction between Si
and bridging oxygen atom.
We hypothesize that the variation of Si-O-Si angle strongly affects the transportation
coefficient of silicates. The decrease of Si-O-Si angle and viscosity of acidic silicate
liquid with increasing pressure is well known experimental fact [2,3,4,5,24,25].These phenomena of silicate liquids
can be explained by weakening of Si-O bonding by bending of Si-O-Si angle, from the view
point of quantum chemical study. The Eyring's viscosity equation is written as follows,
|
η
=
h
N
A
V
m
e
ν
E
β
| (3) |
where h is Plank's constant, N
A is
Avogadro's number, V
m is the volume of molecule, ΔE is the activation energy and
β is the reciprocal temperature. Then we postulate that the molecule in Eyring's viscosity
model of liquid is SiO
4 tetrahedra. The energy change of Si-O bond with varying
Si-O-Si angle cannot be used directly for calculation of the activation energy because the
activation energy expresses not only the exchanging of one bond but also energy barrier of
molecule to slip over the next equilibrium position. The change of bonding energy can affect
to the activation energy because the activation energy is the energy barrier between two
states and the height of energy barrier is affected by the sum of interatomic energy at
intermediate position and the equilibrium position.
4 Conclusion
We here showed the results and discussion of molecular orbital calculations of pyrosilisic
acid molecule. We found the equilibrium geometries for two pyrosilisic acid molecules
(C2V and 60° torsion) using Møller-Plesset perturbation theory and 6-311G (d,p)
split valence basis set. The bent configuration of Si-O-Si angle in equilibrium geometries
can be explained by the balance of Coulombic repulsion between SiO4 tetrahedra
and the energy of lone pair orbitals belonging to bridging oxygen atom without concerning
the contribution of d-p π-bonding. We calculated the energy surface with varying
Si-Obr length and Si-O-Si angle and found the relationship between
Si-Obr length and Si-O-Si bridging angle. The Si-O bond weakens with decreasing
Si-O-Si angle because of instabilization in energy of Si-O bonding orbital with increasing
the hybridization index λ in spλ orbital of bridging oxygen and decrease of
Coulombic interaction between Si and bridging oxygen atom.
References
- 1 G. V. Gibbs, C. T. Prewitt,
K. J. Baldwin, Z. Kristallogr., 145, 108 (1977).
- 2 A. Navrotsky, A. L.
Geisinger, P. McMillan, G. V. Gibbs, Phys. Chem. Miner., 11, 284 (1985).
- 3 E. Ohtani, F. Taulelle, C.
A. Angell, Nature, 314, 78 (1985).
- 4 T. Sakamaki, Y. Wang, C.
Park, T. Yu, G. Shen, J. Appl. Phys., 111, 112623 (2012).
- 5 F. Noritake, K. Kawamura, T.
Yoshino, E. Takahashi, J. Non-Cryst. Solids, 358, 3109 (2012).
- 6 M. D. Newton, G. V. Gibbs,
Phys. Chem. Miner., 6, 221 (1980).
- 7 S. Tsuneyuki, Mol. Eng., 6,
157 (1996).
- 8 J. D. Kubicki, D. Sykes, Am.
Mineral., 78, 253 (1993).
- 9 W. J. Hehre, R. F. Stewart,
J. A. Pople, J. Chem. Phys., 51, 2657 (1969).
- 10 Gaussian 09, Revision A.1,
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.
P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K.
Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T.
Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E.
Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A.
Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M.
Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E.
Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin,
K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A.
D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox,
Gaussian, Inc., Wallingford CT, 2009.
- 11 M. Head-Gordon, J. A. Pople,
M. J. Frisch, Chem. Phys. Lett., 153, 503 (1988).
- 12 Becke, A., D., J. Chem.
Phys., 98, 5648 (1993).
- 13 C. Lee, W. Yang, R. G. Parr,
Phys. Rev. B, 37, 785 (1988).
- 14 J. P. Perdew, K. Burke, M.
Ernzerhof, Phys. Rev. Lett., 77, 3865 (1996).
- 15 A. D. McLean, G. S.
Chandler, J. Chem. Phys., 72, 5639 (1980).
- 16 K. Raghavachari, J. S.
Binkley, R. Seeger, J. A. Pople, J. Chem. Phys., 72, 650 (1980).
- 17 A. Almenningen, O.
Bastiansen, V. Ewing, K. Hedberg, M. Trætteberg, Acta Chem. Scand., 17, 2455 (1963).
- 18 J. P. Foster, F. Weinhold,
J. Am. Chem. Soc., 102, 7211 (1980).
- 19 A. E. Reed, L. A. Curtiss,
F. Weinhold, Chem. Rev., 88, 899 (1988).
- 20 A. E. Reed, F. Weinhold, R.
B. Weinstock, J. Chem. Phys., 83, 735 (1985).
- 21 D. W. J. Cruickshank, J.
Chem. Soc., 1077, 5486 (1961).
- 22 R. S. Mulliken, J. Chem.
Phys., 23, 1833 (1955).
- 23 K. L. Geisinger, G. V.
Gibbs, A. Navrotsky, Phys. Chem. Miner., 11, 266 (1985).
- 24 T. Kushiro, J. Geophys.
Res., 81, 1955 (1976).
- 25 C. M. Scarfe, B. O. Myasen,
D. Virgo, Carnegie Inst. Wash. Yearb., 78, 574 (1979).

