総合論文
核・電子軌道法における原子核軌道エネルギーとプロトン束縛エネルギー計算
2016 年 15 巻 5 号 p. 148-154

詳細
2016 年 15 巻 5 号 p. 148-154
核・電子軌道 (NOMO) 法は,原子核に対して軌道の概念を導入した量子化学理論である.NOMO法におけるHartree-Fock (HF)方程式の解として得られる原子核軌道エネルギーは,全エネルギーや軌道そのものと比較して注目されていない.本論文では,NOMO法における原子核軌道エネルギーの物理的意味とpost-HF理論への応用を述べる.特に,最近開発されたプロトンプロパゲーター法による束縛エネルギーの見積りについて,計算例を含めて紹介する.
量子化学計算では,原子核を点電荷として配置し,電子の波動関数とエネルギーが得られる.原子核の運動は,電子状態計算に由来するポテンシャル超曲面を通して記述される.よく行われる分子の振動解析では,すべての基準振動を調和振動子として近似することで,ポテンシャル超曲面の極小点におけるエネルギーの2階微分から基準振動と振動数が得られる.このような原子核と電子の運動を分離する取り扱いがBorn-Oppenheimer (BO) 近似 [1]であり,Heitler-Londonのモデルによる水素分子の計算 [2]から非経験的かつ定量的な電子状態理論に至るまで,量子化学の基盤として受け入れられてきた.しかし,BO近似に基づいて原子核の量子性を考慮するために,複雑なポテンシャル超曲面上で原子核のSchrödinger方程式を解くのは困難である.
BO近似に依存せずに原子核の量子効果を簡便に考慮できる手法として,核・電子軌道 (NOMO) 法が挙げられる.この手法では,原子核に対して軌道の概念を導入し,電子と原子核の波動関数が同時に決定される.NOMO法はBO近似に基づく分子軌道 (MO) 法と同様に,Hartree-Fock (HF) 近似から出発し [3,4],粒子間の相関を電子相関理論と同様の方法で取り込むことができる [5,6].NOMO法はこれまでに水素結合 [7,8]や二水素結合 [9]に応用され,分子系の構造におけるプロトンの量子効果や同位体効果の説明に成功してきた.
NOMO法では,HF方程式の解として全エネルギー,電子と原子核の軌道,それらに対応する軌道エネルギーが得られる.これまでに全エネルギーの精度向上を目的とした理論開発が盛んに行われてきた.具体的には,粒子間の相関を取り込むためのpost-HF理論への拡張 [5,6],原子核波動関数における並進・回転運動の除去 [4,6,10],あらわな電子-核相関の考慮 [11,12,13]などである.電子-核相関については,密度汎関数理論における相関汎関数として考慮する研究 [7,14]も行われた.
NOMO法と等価な原子核軌道に基づく理論は,他のグループによっても提案され,同様に相関効果や並進・回転運動の除去が検討されてきた [15,16,17,18,19,20,21,22].また,それらの理論を用いて分子系の幾何構造や電子波動関数に対する原子核の量子効果や同位体効果が議論されてきた.このような応用研究は,特に水素結合系に対して盛んである [21,23,24,25].
NOMO法および関連する理論では,系の全エネルギー,電子の軌道および軌道エネルギー,原子核の軌道および軌道エネルギーが得られる.原子核軌道は,配置間相互作用による振動励起波動関数の記述 [26]や,原子核密度を用いた考察 [6,11,12]と関係する.また,位置演算子に関する期待値として原子間距離が得られ,分子系の幾何構造に関する議論に用いられる.これらに対して原子核軌道エネルギーは,これまであまり注目されていなかった.BO近似に基づく電子状態理論の軌道エネルギーに関する研究の進展 [27,28,29,30]を考慮すると,NOMO法における原子核軌道エネルギーについて考察することは意義深い.
本論文の2章では,原子核軌道エネルギーの物理的意味と応用について述べる.3章では,Reyesら [31]によって開発されたプロトンプロパゲーター法に焦点を当て,プロトン束縛エネルギー (PBE) の計算例を紹介する.また,大規模系への応用のために著者ら [32]が開発した分割統治 (DC) 法への拡張についても紹介する.
本節では,NOMO法に対するHF近似であるNOMO/HF法 [3,4]を概説し,原子核軌道エネルギーの物理的意味について述べる.Ne個の電子とNn個の原子核からなる系について,NOMO/HF波動関数は
| (2.1) |
| (2.2) |
| (2.3) |
| (2.4) |
| (2.5) |
| (2.6) |
BO近似に基づくMO法では,Koopmansの定理 [27]が軌道エネルギーに物理的意味を与える定理としてよく知られている.すなわち,占有および非占有軌道エネルギーは,軌道の緩和と電子相関を無視した場合のイオン化ポテンシャル,電子親和力の逆符号にそれぞれ対応する.ここで,NOMO法における原子核軌道エネルギーについて同様に検討する.占有原子核軌道
| (2.7) |
残念ながらKoopmansの定理に基づくPBEは,定性的にも定量的にも満足する結果とならない [31].誤差の原因のほとんどは凍結軌道近似に由来することがわかっている.原子核軌道エネルギーに軌道緩和の効果を考慮することで,PBEの精度を改善することができる.その詳細は,2.3節で説明する.
2.2 NOMO法におけるpost-HF理論と原子核軌道エネルギーNOMO/HF法では粒子間の相関が考慮されていないため,全エネルギーの精度は低い.そこで,配置間相互作用,Møller-Plesset摂動論,結合クラスター法などの理論により相関エネルギーが見積られる.例えば,2次のMøller-Plesset摂動論に基づくNOMO/MP2法の相関エネルギーは [5,6]
| (2.8) |
原子核軌道エネルギーは振動励起の記述にも利用される.配置間相互作用における1粒子励起のみを考慮したNOMO/CIS法 [26]では,励起状態の波動関数は
| (2.9) |
プロトンプロパゲーター法 [31]は,Reyesらによって提案された手法である.これは,多体Green関数が極をもつ条件からプロトンの軌道エネルギーを自己エネルギーで補正する方法である.Reyesらの論文では2次の摂動論に基づくプロトンプロパゲーター (PP2) が報告されている.
NOMO法におけるPP2をNOMO/PP2法と呼ぶ.ここでNOMO/HF波動関数に対するGreen関数G0を考えると
| (2.10) |
| (2.11) |
| (2.12) |
| (2.13) |
| (2.14) |
ωPが求めるべき補正された原子核軌道エネルギーである.右辺第2項と第3項の和が自己エネルギーであり,それぞれ以下の通り与えられる. (2.15) (2.16)
式(2.15)は電子波動関数の緩和と電子-核相関,式(2.16)は原子核波動関数の緩和と核-核相関からなる.自己エネルギーはωPを含むため,式(2.14)は繰り返し計算で解く.
NOMO/PP2法の利点は,すべてのプロトンの束縛エネルギーを1度のNOMO/HF計算から得られることである.すなわち,多数のプロトンを有する系に対して,PBEの傾向を容易に調べることができる.また,電子相関エネルギーの線形スケーリング手法である分割統治 (DC) 法 [33,34,35]により生成される局所軌道を用いることで,計算コストを線形スケーリングにできる.この方法をNOMO/DC-PP2法 [32]と呼ぶ.
DC法では系を部分系に分割し,その周囲を緩衝領域として指定し,両者をあわせて局在化領域と呼ぶ.DC法に基づく電子相関エネルギーの計算では,局在化領域の分子軌道からエネルギーを計算し,エネルギー密度解析 [36]に基づいて部分系の相関エネルギーを得る.一方,NOMO/DC-PP2法では,Figure 1のように注目する水素原子を部分系とし,その周囲に緩衝領域を指定する.そして,Fock行列の対角化によって得た局在化領域の軌道をそのまま式(2.15)に適用すればよい.

Schematic illustration of the subsystem (orange), buffer region (blue), and localization region (orange + blue) in the NOMO/DC-PP2 method.
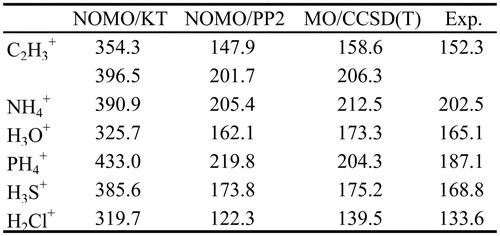
本節では,NOMO/PP2法によるPBE計算の例として,6種類のカチオンへの適用を紹介する.計算はGAMESS [37]に実装したルーチンを用いて行った.基底関数として,電子に対してaug-cc-pVTZ,プロトンに対して(3s3p3d)を使用し,プロトン以外の原子核はすべて点電荷とした.プロトンに対する基底関数の指数は,even-temperedのスキーム [4,38]に基づいて決定した.カチオンの構造は,B3LYP/6-31G (d)レベルの最適化構造である.比較対象として,Koopmansの定理に基づく見積り,BO近似に基づくCCSD (T)/aug-cc-pVTZレベルの電子エネルギー差を掲載した.以後,前者をNOMO/KT,後者をMO/CCSD (T)と表記する.MO/CCSD (T)の計算で用いた構造はNOMO法と同一であり,ゼロ点振動エネルギーは含まない.さらに,中性分子のプロトン親和力 [39]を,カチオンのPBEの実験値として掲載した.
Table 1より,NOMO/KTはPBEを大幅に過大評価することがわかる.これは,プロトン脱離後の電子波動関数の変化が考慮されていないためである.NOMO/PP2法ではこの状況は改善され,MO/CCSD (T)に近い結果が得られた.NOMO/PP2法とMO/CCSD (T)の差異は,主にプロトンプロパゲーター法の高次の項に由来すると考えられる.NOMO/KTのMO/CCSD (T)からの差が最大であるPH4+に注目すると,NOMO/PP2法によるPBEがMO/CCSD (T)より約15 kcal/mol大きく,高次の影響が大きいと推測される.また,MO/CCSD (T)と実験値の差は,ゼロ点エネルギーとプロトン脱離後の分子構造の緩和が主な原因と考えられる.NOMO/PP2法によるPBEを実験値と比較すると,C2H3+,NH4+,H3O+,H3S+,H2Cl+では,15 kcal/mol以内の差である.C2H3+では,プロトン間の束縛エネルギーの違いも再現している.それに対してPH4+では30 kcal/mol以上の過大評価となる.このようにNOMO/PP2法によるPBEはおおむね妥当な挙動を示すが,実験値と比較する際には考慮されない効果に注意する必要がある.
本節では,NOMO/DC-PP2法を用いたPBEの計算例を紹介する.解離しうるプロトンを多数含む分子として,ポリフェノールの一種であるテアフラビン-3,3'-ジガレートを計算対象とした.ωB97X-D/6-31G (d,p)レベルの量子化学計算により最適化された構造をFigure 2に示す.Figure 2における13個のフェノール性ヒドロキシ基に注目し,NOMO/DC-PP2法によるPBE計算を行った.基底関数は電子に対して6-31G (d,p),OH基のプロトンに対して(3s3p3d)を使用した.それ以外の原子核はすべて点電荷として扱った.プロトンに対する基底関数の指数は,前節と同じ値を用いた.部分系は水素原子,緩衝領域はプロトンの基底関数中心から2 Å ∼8 Å以内の原子とし,PBEの緩衝領域に対する依存性を調べた.

Geometry of theaflavin-3,3′-digallate for the NOMO/DC-PP2 calculation. Hydrogen atoms except for hydroxy groups are not described.
計算結果をFigure 3にまとめた.緩衝領域がプロトンから2 Åの場合は,4 Å以上の場合とPBEの傾向が全く異なる.一方,6 Åと8 Åの結果はほぼ一致しており,PBEが緩衝領域のサイズに関して収束することがわかる.この結果は,PBEにおける電子分布の緩和や相関効果が局所的であるためと考えられる.緩衝領域が4 Å以上の場合では,1番から4番までのプロトンは順番に束縛エネルギーが減少する.これら4つのプロトンは水素結合に関与しており,プロトン解離後の負電荷と周囲のヒドロキシ基の静電相互作用を考えると,妥当な傾向と言える.同様の関係は,5番と6番,7番から9番,11番と12番の間でもみられる.

Buffer-size dependence of the NOMO/DC-PP2 method. The numbering of protons corresponds to Figure 2.
以上の結果は孤立分子の平衡構造に対して一点計算を行ったものである.周囲に水分子をあらわに配置した分子動力学シミュレーションを行い,複数のスナップショットに対してNOMO/DC-PP2計算を行うことで,水溶液中でのプロトン解離能をPBEに基づいて議論できる.
本論文では,NOMO法における原子核軌道エネルギーの物理的意味と,それに基づくPBEの見積りについて述べた.原子核軌道エネルギーは,NOMO法におけるpost-HF計算に用いられるだけでなく,Koopmansの定理により意味付けられる.すなわち,占有原子核軌道エネルギーの逆符号は,凍結軌道近似のもとで原子核を系から取り除くエネルギーに相当する.
凍結軌道近似で考慮されない電子分布の緩和を考慮した手法がプロトンプロパゲーター法である.これにより,PBEの計算精度が大幅に改善されることが数値的に示された.さらにDC法との組み合わせにより,多数のプロトンに対する束縛エネルギーが容易に得られるようになった.
BO近似に基づくMO法では,系内に存在するすべてのプロトンに対して束縛エネルギーを求める場合,その数だけ脱プロトン状態の計算が必要となる.一方,本稿で紹介したNOMO/DC-PP2法では,1度の計算によりすべてのプロトンに対して束縛エネルギーを求めることができる.これまでNOMO法に関する研究は,非断熱効果や原子核の量子効果などBO近似に基づくMO法では直接記述できない現象を対象としてきた.しかし,NOMO/DC-PP2法は,MO法でも取り扱いが可能なPBEの計算を対象としており,より効率的な計算手法という位置づけである.NOMO法の開発に長年携わってきた著者らにとっては,この新しい展開に大いに期待を寄せている.
本研究は,文部科学省・科学研究費補助金(基盤A No. 26248009「ユビキタス水素の機能とダイナミクスに関する理論的研究」),独立行政法人科学技術振興機構・戦略的創造研究推進事業(CREST)「相対論的電子論が拓く革新的機能材料設計」,文部科学省・元素戦略研究拠点「実験と理論計算科学のインタープレイによる触媒・電池の元素戦略研究拠点(ESICB)」の支援を受けて実施された.また,量子化学計算の一部は自然科学研究機構(NINS)・計算科学研究センター(RCCS)の計算機を利用して行った.