研究論文
化合物のAmes予測におけるGraph Convolutional Networkの特徴評価
キーワード:
Graph Convolutional Network,
Machine learning,
Ames test,
Applicability Domain,
Structural similarity
2021 年 20 巻 1 号 p. 1-9
詳細
2021 年 20 巻 1 号 p. 1-9
医薬品候補物質の潜在的な発がん性早期警戒システムであるAmes試験のin silico予測は,創薬研究において重要な予測項目の一つである.in silico予測の一手法である機械学習による予測では,Applicability Domain (AD)という機械学習モデルが本来の性能を発揮できるデータ領域を定義する研究がある.創薬研究においては,学習データと構造類似性が低い医薬品候補化合物の予測を行う場合があり,そのような化合物はAD領域外になる可能性が高く予測精度が低下する傾向がある.本研究では,Ames試験の機械予測モデルを作成し,テストデータとしてAD領域内/外となる確率が高い化合物群をそれぞれ用意して,複数の機械学習手法の予測性能を評価した.人工知能技術の発展により,創薬分野でも注目を集めているGraph Convolutional Network (GCN)と既存の機械学習手法の予測性能を比較した結果,AD領域外となる可能性が高い化合物群の予測性能において,GCNは既存手法より優れていた.
Ames test to detect mutagenicity in vitro is of crucial importance in drug discovery and development as an early alerting system for potential carcinogenicity and/or teratogenicity for drug candidates. In the alerting system of machine learning approaches, which are the main approach of in silico prediction, there is a concept called Applicability domain (AD) that has a significant influence on prediction accuracy. In drug discovery, prediction of drug candidate compounds with low structural similarity to the training data may take place, while such compounds have a high probability of being out of the AD and tend to be less accurate. In this study, we evaluated the performance of several machine learning methods for a group of compounds with a high probability of being included in AD or not, respectively. The results showed that the performance of the graph convolutional networks (GCN) method which have achieved a superior performance in a wide range of tasks was better than conventional methods. In particular, the accuracy of the GCN method is significantly different from the conventional methods for the prediction of molecules with low structural similarity (Figure 4).
Ames試験は,化合物の潜在的な発がん性,催奇形性の早期警戒システムとして,創薬において重要な試験項目の一つである.in silicoによるAmes試験予測は,ICH M7「潜在的発がんリスクを低減するための医薬品中DNA反応性(変異原性)不純物の評価及び管理」ガイドライン [1]において,リスク評価に用いることが可能と言及されており,創薬研究においてAmes試験の予測精度向上,予測精度評価は重要である.
代表的なin silico予測手法の一つである機械学習において,Zhuらは,機械学習モデルが本来の性能を発揮できるデータ領域をApplicability Domain (AD)と定義して,その領域内の化合物の予測精度は,領域外の化合物の予測性精度より高いことを報告している [2].Zhuらは,また,予測対象化合物がAD領域外となる大きな理由として,構造類似度が影響していることを報告しており,予測対象化合物が学習データと類似しているほど,AD領域内になる可能性が高くなることを示唆している.しかし創薬研究においては,着目する医薬品候補化合物と類似した構造が学習データに含まれないことが多く,医薬品候補化合物がAD領域外となる傾向がある.機械学習によるAmes試験の予測において,このような医薬品候補化合物の予測精度は課題である.
一方,化合物の機械学習手法は,情報処理分野における人工知能(AI; Artificial Intelligence)技術の発展とともに,新しい手法が提案されており,その一つがグラフ畳み込みネットワーク(GCN; Graph Convolutional Network)である [3].機械学習手法の一つであるディープラーニング [4]は,画像処理分野で大きな成果を挙げているが,その成果への貢献が大きい手法の一つが,畳み込みニューラルネットワーク(CNN; Convolutional Neural Network)である [5].畳み込みとは,関数を平行移動しながら重ねて足し合わせる二項演算で特徴抽出に適した手法であり,画像データにおいては威力を発揮する.グラフ構造,すなわち,ノード(頂点)群とノード間の連結関係を表すエッジ(枝)群で構成される抽象データ型に対して畳み込みを行う手法の一つが,Graph Convolution Network (GCN)である.原子をノード,原子の結合をエッジとすれば,化学構造式はグラフ構造であるため,適切に化学構造を表現できると考えられGCNを適用した機械学習の応用が期待されている.
化学構造を表現する方法の代表的手法は,フィンガープリント法であるが,これは分子の局所的な構造を考慮した特徴表現手法であり,近傍の定義を事前に定める必要がある [6].一方,GCNによる化学構造表現は,静的距離の近傍の原子だけでなく,畳み込みにより,遠方の原子の情報を動的に反映することができる.また,フィンガープリントでは,単一の原子特性(原子種,もしくは,水素結合性等の特徴)と原子間の結合の種類から表現されるが,GCNでは原子に複数の特性を持たせる表現が可能である.
GCNを化学構造の実験値予測に応用した例では,Liらの化学的安定性 [7],Ishidaらの合成反応予測に用いた報告 [8]がある.毒性予測に応用した例では,Wuらは [9]複数のターゲットの薬理活性,溶解度や脂溶性等の物性,複数の毒性予測について多くの手法の比較結果を報告している.Wuらの報告では,GCNの有用性が示唆されているものの,各予測対象の学習データ,テストデータは固定であるため,どのような学習データを用いた場合に予測性能が優れているか,適用範囲が明らかでない.
そこで,本研究では,GCNを化学構造に応用した場合,どのような条件下で既存手法より予測性能が向上するかを明らかにするために,Ames試験について,フィンガープリント法と代表的な化合物のクラス分けで用いられる既存の3手法(Support Vector Machine,Random Forest,Gradient Tree Boosting)の組み合わせによる予測精度と,GCNの予測精度を比較した.特に,AD領域外の化合物に対する予測性能について比較するために,テストデータとして学習データと類似している化合物群と類似してない化合物群を用意し,それぞれに対する予測精度を評価し,各手法に与える影響について考察した.
手法評価には,Hansenらの論文 [10]のAmesの実験データセットを用いた.このデータセットは,CCRIS [11],複数の論文(Helmaら [12],Kaziusら [13],Fengら [14]),VITIC [15]),及びGeneToxデータセット [16]から得ており,機械学習用のデータとして整備済みである.その主な処理内容は次の通りである.
・重複構造を削除
・異常および無機分子構造を削除
・DEREK [17],または,MULTICASE [18]の予測モデル作成に用いられたデータに対して矛盾する実験結果である化合物を削除
最終的なデータセットの分子の数は,陽性3,503個,陰性3,009個,計6,512個である.我々は,KNIME [19]のRDKit [20]ノードを用いて,このデータセットを,本研究の評価用に再処理を行った.その際,6化合物は,RDkitにおいてエラーが発生したため,本研究で解析に用いた化合物数は6,506個であり,陽性化合物3,497個,陰性化合物3,009個の内訳であった.
我々は,このデータを,学習用データとテスト用データに分割するにあたり,2つの方法で行った.一つ目の方法では, Hansenらがstaticと定義したデータセットを学習データとして,残りのデータをテストデータとした.staticとは,DEREK,MULTICASEの双方において,信頼性が高いと判断できた既知化合物群である.二つ目の分割方法では,全化合物からランダムに,学習データとテストデータに分割した.その際,学習データの総数,陽性,陰性化合物数がそれぞれ上記のstaticと同じ数となるように分割した.さらに,ランダムに分割したデータセットは,乱数の設定を変えた2種類のデータを作成し,計3つのデータセット(それぞれ,Static,Random1,Random2とする)を作成した.
化合物の構造類似性による予測精度の違いを観察するために,それぞれのデータセットのテストデータを,学習データに類似している化合物とそれ以外の化合物に分割した(Figure 1).化合物の類似度は,RDKit Morgan Fingerprint (1024bit; radius=3)を用いたTaniomoto係数により算出し,0.5を閾値として,学習データに対する最近傍類似度(closest similarity)が0.5より大きい値を類似している化合物セット(テスト-類似, Test-sim)とし,それ以外の化合物を類似してない化合物(テスト-非類似, Test-disim)とした.データセットの内訳をTable 1に示す.

The workflow of splitting test data.
| Static data set | Random1 data set | Random2 data set | |||||||
| Trainingdata | Test-sim | Test-disim | TrainingData | Test-sim | Test-disim | Trainingdata | Test-sim | Test-disim | |
| Pos | 951 | 400 | 1,658 | 951 | 604 | 1,454 | 951 | 614 | 1,444 |
| Nega | 634 | 507 | 2,356 | 634 | 921 | 1,942 | 634 | 903 | 1,960 |
| 1,585 | 907 | 4,014 | 1,585 | 1,525 | 3,396 | 1,585 | 1,517 | 3,404 | |
予測精度の評価は,各予測モデルのProbabilityによるROC曲線(Receiver Operating Characteristic curve)のAUC (Area Under the Curve)で行った.ROC曲線は,予測性能を2次元のグラフで表したものであり,縦軸に真陽性率,横軸に偽陽性率をプロットして線で結ぶ.このグラフの下の部分の面積がAUCの値で0から1までの値をとり,値が1に近いほど予測精度が高いことを示す.
2.3 Graph Convolutional NetworkGCNのネットワーク構成は,入力層,畳み込み層,プーリング層,集約層からなり,各層の概要は次のとおりである.入力層では,指定された化学構造のグラフ情報と,各原子のプロパティのベクトルにより入力値が作成される.畳み込み層では,任意の原子に対して,隣接している原子の特徴ベクトルを畳み込み関数により算出する.この処理を全原子に対して繰り返し行う事で,隣接している原子の特徴が反映されたベクトルが作成される.プーリング層では,畳み込み層で算出された特徴ベクトルの各要素について,最小値等の代表値を算出する.この処理では,代表値を残し他のデータは破棄するため,情報量を下げてグラフ構造の特徴を際立たせる処理となる.集約層では,畳み込み層,プーリング層で算出された各原子の特徴ベクトルを,要素ごとに加算して化合物の特徴ベクトルを算出する層である.
GCNは,ディープラーニングの一手法であり,各層において,ディープラーニングの大きな特徴の一つである,ドロップアウトアルゴリズムが適用可能である.ドロップアウトとは,各層のニューロンを定めた確率により働かなくすることで,意図的に情報を欠損させた状態で学習させる.そのような状態でも予測性能を維持するモデルを作成することで,機械学習の大きな課題である過学習を防ぎ,予測モデルの頑健性を高める手法である.
本研究では,DeepChem [21]を用いてGCNの予測モデルを作成しており,各原子の特性は{原子種,原子が結合している水素以外の原子数,データ上で明示されてない結合水素数,原子のフォーマルチャージ,ラジカル電子数,軌道の種類,芳香環に含まれるか否か,総結合水素数(データ上の明示,非明示双方)}の8種類である.予測モデルを作成する際には,多くのハイパーパラメータ設定が必要となるが,本研究では,畳み込み層を{64要素*2階層,64要素*3階層,64要素*4階層}(それぞれDeepChemの表記で,[64, 64], [64, 64, 64], [64,64,64,64])の3パターン,ドロップアウト確率を{0.2, 0.5, 0.9}の3パターン,学習回数であるエポック数は200に固定し,計9パターンの組み合わせで予測モデルを作成し,各テストセットに対するROCの最大値をGCNの予測値として採用した.その他のハイパーパラメータはデフォルト設定を用いた.
2.4 Support Vector Machine (SVM)SVMアルゴリズムは,Ivanciucらによって開発された [22].SVMアルゴリズムの大きな特徴は,マージン最大化とカーネルトリックの導入である.SVMでは,各サンプルはベクトルで表現されるが,サポートベクトルと呼ばれるクラス境界近傍に位置するベクトルをクラス分けの基準とする.この際,その距離が最も大きくなるような位置に識別境界を設定する.マージン最大化とは,マージンと呼ばれるこの距離を最大化するように学習を行う手法である.カーネルトリックとは,SVMの予測性能を高めたアルゴリズムであり,線形空間では分離が難しいデータを非線形空間で行う手法である.本研究では,SVMをscikit-learn [23]で実装し,カーネルはデフォルトのRBFカーネルを用いた.
RBFカーネルを用いた学習時には,CostとGammaのハイパーパラメータチューニングを行う.Costは誤分類をどの程度許容するかのパラメータであり,Costが大きいほど誤分類を許容せず,学習データへフィッティングしやすくなる.Gammaは境界の複雑さであり,大きくなるほど境界は複雑となる.本研究では,各データセットに対して,Costを{1, 10, 100, 1000},Gammaを{1.0, 0.1, 0.01, 0.001}の総当たりの組み合わせによる16個の予測モデルを作成し,テストセットそれぞれに対して最も予測精度が高い結果を予測性能として採用した.
2.5 Random Forest (RF)RFは,複数の決定木予測モデルを組み合わせてより強力な予測モデルを作るアンサンブル学習手法の一つである.決定木とは,条件に基づいてデータを分割していく学習方法で,「ある変数について,ある閾値で予測結果が分岐する」ということを繰り返して予測モデルを構築する.
RFのアンサンブル学習は,学習データセットから重複を許してサンプリングし,そのサンプル集合に対して決定木の作成を繰り返し複数の決定木を作成する.これら複数の決定木はそれぞれ独立しており,予測結果は,作成された決定木の予測結果の平均となる.
本研究では,決定木の階層数を{5, 7, 10, 15}で試行し,アンサンブルの数は100に固定,scikit-learnにより各データセットに対し4個の予測モデルの作成を行った.決定木の分岐に用いられる変数の選択方法である重要度指標は,デフォルト設定であるGini indexを用い,各テストセットの最も予測精度が高い結果を,予測性能として採用した.
2.6 Gradient Tree Boosting (GB)GBもRFと同じく決定木ベースのアンサンブル学習の一つである.GBとRFの大きな違いは,GBではブースティングという手法により,複数の決定木を作成する.ブースティングでは,まず,基本となる決定木予測モデルを作成してベースラインとする.その後は,この基本モデルの間違った予測に焦点を当てて,間違った予測を修正するように「重み」を加味して次の予測モデルを作成することで,予測結果を改善していく.これを繰り返し行い,最終的に全ての決定木の結果から予測を行う.
予測木を作成する時のチューニングパラメータとして,決定木の階層数を{5, 7, 10, 15},学習レートを{0.1,0.05,0.01}の組み合わせを全て試行した.予測モデルの作成は,scikit-learnにより行い,その他のパラメータはデフォルト値を用いた.各テストデータあたり12個の予測モデルを作成し,その中で最も予測精度が高い結果を採用した.
2.7 ディスクリプタSVM,RF,GBにおいては,予測を行うために化合物を表現するディスクリプタが必要となる.本研究では,データセット作成の類似度計算に用いたフィンガープリントをディスクリプタとした.フィンガープリントは1024ビットで作成しているため,入力ディスクリプタの数は1024個である.
各手法の予測精度(ROCのAUC)をTable 2に,そのグラフをFigure 2に示す.また,GCNと既存手法との予測精度の差をTable 3に示す.GCNと既存手法との予測精度の差の最小値を確認すると,Staticデータセットのテスト-類似では,0.0142,テスト-非類似では0.0948であった.同様にRandom1データセットにおいては,それぞれ,0.0086,0.0186,Random2データセットでは,0.0093,0.0489であった.このように,GCNの予測精度は全てのケースにおいて,差が正の値となり,既存手法より優れていた.
| Static data set | Random1 data set | Random2 data set | ||||||||||
| GCN | SVM | RF | GB | GCN | SVM | RF | GB | GCN | SVM | RF | GB | |
| Test-sim | 0.827 | 0.790 | 0.806 | 0.813 | 0.895 | 0.876 | 0.874 | 0.886 | 0.890 | 0.868 | 0.878 | 0.881 |
| Test-disim | 0.764 | 0.668 | 0.667 | 0.670 | 0.813 | 0.771 | 0.780 | 0.794 | 0.823 | 0.774 | 0.768 | 0.774 |
| difference | 0.0631 | 0.1220 | 0.1394 | 0.1437 | 0.0817 | 0.1051 | 0.0942 | 0.0918 | 0.0675 | 0.0946 | 0.1102 | 0.1072 |
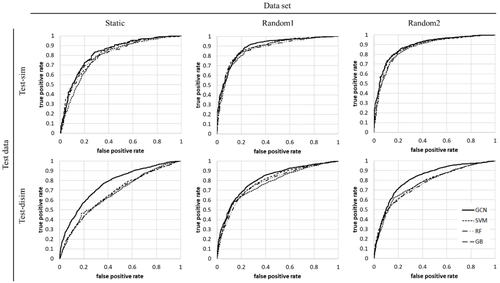
The ROC curves of each machine learning method. The AUC of each plot were shown in Table 2.
| Static data set | Random1 data set | Random2 data set | |||||||
| SVM | RF | GB | SVM | RF | GB | SVM | RF | GB | |
| Test-sim | 0.0374 | 0.0213 | 0.0142 | 0.0184 | 0.0205 | 0.0086 | 0.0223 | 0.0127 | 0.0093 |
| Test-disim | 0.0963 | 0.0976 | 0.0948 | 0.0417 | 0.0329 | 0.0186 | 0.0494 | 0.0554 | 0.0489 |
テストデータの構造類似性に着目すると,どの手法においてもテスト-類似に対する予測のほうが,テスト-非類似に対する予測より優れていた.ZhuらのADの報告 [2]では,学習セットとの構造類似度が高いテスト化合物群は,低いテスト化合物群よりAD領域内となる構造が多い.このことから,テスト-類似はAD領域内になる確率が高い化合物群であり,本研究においても,先行研究と同様にAD領域内のテストデータのほうが,AD領域外のテストデータより予測性能が優れる傾向を示した.この傾向は,GCNと既存手法で共通であった.
予測精度が最も悪かったのは,どの手法においても,Staticデータセットのテスト-非類似化合物群に対しての予測であった.このデータセットでは,分子量250以下の化合物の割合が,学習データでは約70%であるのに対して,テストデータでは55%と分子量分布が異なっている(Figure 3).さらに,学習データと構造類似度も低いことから,学習データに含まれてない部分構造がテストデータに存在していると考えられる.一方,Random1・2では,全体の化合物群から無作為に学習データとして選択しているため,テストデータを分割した構造類似度は0.5でStaticデータセットと変わらないにも関わらず,分子量分布は学習データと類似している.

The histogram of the molecular weight of each data set.
Table 4は,学習データに含まれてない部分構造を確認するために,各データセットの化合物のフィンガープリントビットのうち,出現回数が1%以上のフィンガープリントビット(メジャーフィンガープリントビットと呼ぶ)を集計した表である.Staticデータセットでは,学習データにはなく,テストデータのみにあるメジャーフィンガープリントビットは120個であったが,Random1・2のデータセットでは,それぞれ10,18個と少なかった.また,学習データとテストデータすべてに存在していたメジャーフィンガープリントビットは,Static,Random1・2の各データセットにおいて,それぞれ,738,895,900個でありStaticデータが最も少なく,StaticデータとRandom1・2で異なる傾向であった.
| condition1 | condition 2 | Static data set | Random1 data set | Random2 data set |
| not exist in training set | only exist in test-sim | 3 | 2 | 1 |
| only exist in test-disim | 92 | 22 | 23 | |
| exist in both | 120 | 10 | 13 | |
| exist in training set | only exist in test-sim | 1 | 16 | 2 |
| only exist in test-disim | 67 | 67 | 76 | |
| exist in both | 738 | 895 | 900 | |
| else | 3 | 12 | 9 | |
予測精度は,全ての手法において,StaticデータセットよりRandom1・2のデータセットのほうが優れており,Staticデータセットでは,学習データに含まれてないメジャーフィンガープリントビットがテストデータに多く含まれていたことが,他のデータセットより予測精度が低くなった原因と考える.各データセットの類似/非類似の分割に用いた類似度閾値は同じであるため,予測性能に与える影響は,分子全体の類似度より,テストセットに含まれていないメジャーフィンガープリントビット(≒部分構造)の影響が大きいといえる.
3.2 GCNと既存手法との比較各手法における,テスト-類似,非類似の予測精度の差を確認すると,既存手法では,最小0.095,最大0.144であったのに対して,GCNは最小0.063,最大0.082と,いずれも既存手法より差が小さかった.このことから,GCNは,学習データと構造類似度が低くAD領域外になる確率が高い化合物群の予測性能が,既存手法より,より優れていた.
特に,Staticデータセットのテスト-非類似の予測精度は,GCNと既存手法の差が最も大きかった.各テストデータに対する既存手法とGCNのROC散布図(Figure 4)を見ると,プロット点はROC AUCが小さいほど直線から離れている傾向であり,既存手法での予測精度が悪いほど,GCNの予測精度がより優れている傾向であった.

The scatter plot of GCN vs conventional methods. (dot line is the equality line; dot circle is the group with the largest difference)
すなわち,GCNは,学習データと構造類似度が低い化合物群の予測性能が既存手法より優れており,特に,学習データには少ない部分構造を持つ化合物群において,より,既存手法より予測性能が優れている特徴を持つ.
一方,テスト-類似に対する予測精度は,Random1・2のデータセットにおいて,既存手法とGCNと予測精度の差が小さく,学習データと類似している構造群に対する予測性能は,GCNのほうが優れていたものの,既存手法と大きな違いはないと考える.
3.3 既存手法間比較GCN以外のSVM,RF,GB,各手法間の予測結果を比較すると,ROCの差は最大で0.023,ほとんどの場合0.015未満であり,GCNとの差に比べると小さく,既存手法でディスクリプタをフィンガープリントとした場合,既存手法間の差は小さかった.
3.4 GCNのハイパーパラメータと予測精度の関連予測精度に影響を与えるハイパーパラメータの効果を確認するために,既存手法との差が大きかったStaticデータセットのテスト-非類似に対する予測精度を,全ハイパーパラメータの組み合わせについてTable5にまとめた.ドロップアウトは頑健な予測モデル作成に影響するハイパーパラメータであるが,ドロップアウト0.9の場合,64*3,64*4階層において学習が収束しなかった.これは情報が欠損しすぎて学習できなかったと推察する.ドロップアウト0.5の64*3階層の時には,最も良い予測精度に近い0.762の値をとり,同じドロップアウトの64*4,64*2階層も0.75を超える良い予測精度と.ドロップアウト0.5が総じて良い予測精度であった.しかし,最も良い予測精度であったのは,ドロップアウトが0.2;畳み込み層が64*2階層の時であり,ROC AUCは0.764であった.AD領域外となる可能性が高い化合物群の予測精度が既存手法より優れていた理由がドロップアウトの効果であるか否か,また,畳み込み層の構造が影響しているかは,不明瞭な結果であった.

Stevenらは,グラフ構造による化合物表現では,ディープラーニングのネットワーク構造によりフィンガープリントに比べて化合物構造表現の柔軟性が異なることを報告している [24].また,グラフ構造による化合物表現は予測モデルと一体化しており,フィンガープリントと予測モデル作成手法の関係のように明確な区別がない統合的なアプローチであることを述べている.本研究においては,ディープラーニング系の特徴であるハイパーパラメータ(ドロップアウト,畳み込層の構造)の影響は明らかではなく,学習が収束したどの組み合わせでも0.7以上の予測精度であった.このことから,GCNの予測精度が優れていた理由として,ディープラーニングによる予測モデル作成の影響よりも,化合物をグラフ構造で表現し予測モデルと統合することで化合物構造表現が柔軟になるGCNの特徴が,既存の予測モデルより予測精度が優れていた理由と考える.
本研究では,化学構造からAmesの実験結果を予測する手法について,複数の予測手法を比較した.GCNは,本研究で作成した全てのテストデータセットに対して,最も優れた予測精度であった.また,GCNは,AD領域外となる可能性が高い化合物群に対して,より優れた予測性能を示した.特に,学習データセットに少数しか含まれてない部分構造を含むような化合物群の予測において,既存手法より優れていた.創薬研究においては,医薬品候補化合物を特許・権利化するために,化学構造の新規性が求められる.その結果,学習データセットには含まれない部分構造を持つ化合物のAmes予測が求められることがある.本研究で示したGCNの特性は,そのような場合において,既存手法より有効であると考える.GCNが優れていた理由は,ドロップアウト,畳み込み層のハイパーパラメータの影響は明確ではなく,GCNのグラフ構造と予測モデルの統合的アプローチであると考えた.
本研究においては,脂溶性や分子量に代表される分子全体を表現する情報(分子ディスクリプタ)を用いず,化学構造のみで表現できる情報を用いて予測を行っており,分子ディスクリプタを使わない場合,GCNは既存手法より優れていると考えられる.今後は,既存手法において,フィンガープリントだけでなく,分子ディスクリプタも利用した場合にGCNの優位性が保たれるかについて研究を進めたい.また,Ames試験以外の予測においても,本研究で実施した方法と同様のデータ分割を行うことで,GCNがAD領域外となる可能性が高い化合物群の予測精度を考察できると考える.