Letters (SCCJ Annual Meeting 2022 Spring Poster Award Article)
Improvement of Interatomic Potential for Molecular Dynamics Simulation of β-LiAlSiO4
2022 Volume 21 Issue 2 Pages 33-35
Details
2022 Volume 21 Issue 2 Pages 33-35
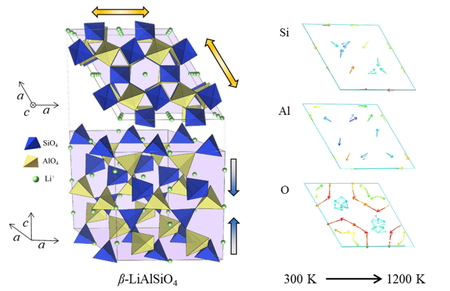
Thermal change of lattice parameters of β-LiAlSiO4 crystal simulated by molecular dynamics simulation was improved by revision of the interatomic potential. The discontinuity of thermal change of c-axis lattice parameter observed in the previous work between 800 K and 900 K was dissolved, but the simulated linear thermal expansion of c-axis was smaller than the reference data. The visualized shift of relative coordinates of each atom with the temperature increase from 300 K to 1200 K showed the different variation between the two types of double helix structures that exist in the unit cell.