Abstract
Metalloproteins such as 4-hydroxyphenylpyruvate dioxygenase (HPPD) and Cytochrome P450 are important for agrochemical research. In classical mechanics, to reproduce the metal-coordination environment of metalloproteins, various models (Nonbonded model, Bonded model, Nonbonded/Bonded Hybrid model etc.) for metal complexes have been developed. In this study, we built force field parameters for the molecular dynamics simulation of HPPD and evaluated the Fe2+-coordination environment. The result revealed that the hybrid model is suitable for the simulation of HPPD and able to simulate the metal-ligand (water) exchange process.
Translated Abstract
Metalloproteins such as 4-hydroxyphenylpyruvate dioxygenase (HPPD) and Cytochrome P450 are important for agrochemical research. In classical mechanics, to reproduce the metal-coordination environment of metalloproteins, various models (Nonbonded model, Bonded model, Nonbonded/Bonded Hybrid model etc.) for metal complexes have been developed. In this study, we built force field parameters for the molecular dynamics simulation of HPPD and evaluated the Fe2+-coordination environment. The result revealed that the hybrid model is suitable for the simulation of HPPD and able to simulate the metal-ligand (water) exchange process.
1 緒言
分子動力学計算 (MD) は, タンパク質の動的挙動解析やタンパク質と阻害剤間の相互作用解析等に利用され, 創農薬研究でも欠かせない技術の一つである. 創農薬研究で標的とするタンパク質は, 金属含有酵素であることも多い. 創農薬研究において, 金属含有タンパク質の妥当なMDを実行するためには, 金属活性中心の配位環境を古典力場で正しく再現する必要がある. これまでに, 金属活性中心の配位環境の古典力場での取り扱い方法としては, Nonbondedモデル[1], Bondedモデル[2]およびNonbonded/Bonded Hybridモデル[3]等が開発されている. そこで, 本研究では, 除草剤標的として知られる非ヘム鉄含有タンパク質である4-hydroxyphenylpyruvate dioxygenase (HPPD) を対象に, 鉄配位環境の力場パラメータを構築し, HPPD阻害剤の一種であるメソトリオンとの結合を再現できるモデルの探索を行った.
2 方法
シロイヌナズナHPPDのアポ体 (PDB: 1SQD) およびメソトリオン複合体 (PDB: 5YWG) の活性部位を抜き出し, クラスターモデルを構築した (Figure 1). 古典力場計算には, Ambertools21を利用し, 量子化学計算にはGaussian16 Rev. Cを利用した. 力場パラメータとして, アミノ酸およびメソトリオンには, Amber ff14SBおよびGAFF2を割り当て, 水分子はTIP3Pモデルで取り扱った. 力場パラメータおよびRESP電荷の算出はB3LYP/6-31*レベルで行った.
本研究では, (i) Nonbondedモデル, (ii) Bondedモデル, (iii) Hybridモデルの3種類を準備した.
Nonbondedモデルにおける鉄は, Li/Merzのパラメータ[1]を利用した. Bondedモデルにおける鉄とその配位子間の結合や角度に関連する平衡値, 力の定数およびRESP電荷は, MCPB.py[4]で求めた. Hybridモデルでは, アミノ酸残基と鉄との間はBondedモデルで取り扱い, メソトリオンおよび水と鉄との間はNonbondedモデルで取り扱った. Hybridモデルにおける鉄とアミノ酸残基との結合や角度に関連する平衡値, 力の定数およびRESP電荷はアポ体を用いてMCPB.pyにより構築した. 鉄とアミノ酸残基との総和形式電荷を+1.0とするため, 三つの配位水に分布するRESP電荷を各アミノ酸残基の主鎖N原子上の水素原子およびCα原子上の水素原子に割り当て, 系の電荷調整を行った.
構築した力場パラメータを利用して, アポ体および複合体に関して真空中で構造最適化を行った. また, 古典力場における結果と比較するために, B3LYP/6-31G*レベルで, アポ体および複合体に対して真空中で構造最適化を行った.
次に上記で構築したNonbondedモデルおよびHybridモデルの力場パラメータを利用して, シロイヌナズナHPPDアポ体に対する100 nsのNPTシミュレーション (T = 300 K) を実施した. 初期構造は, シロイヌナズナHPPD-メソトリオン複合体 (PDB: 5YWG) を基に, メソトリオンの除去および1SQDからの結晶水の挿入により作成した. クラスターモデルにおける解析の結果, Bondedモデルは鉄配位環境の再現に問題があったため, Bondedモデルを用いたMDは実施しなかった. HPPDには, Amber ff14SBを割り当て, 水分子はTIP3Pモデルで取り扱った. MDには, Gromacs2019.6を利用した.
3 結果
鉄-配位子間の結合距離および配位子-鉄-配位子間の結合角度について, 最適化構造とX線結晶構造との平均絶対誤差 (MAE) を求めた (Figure 2). Nonbondedモデルでは, B3LYP/6-31G*の結果と比較すると, アポ体および複合体の両者で結合距離では同程度の誤差であり, 結合角度ではやや誤差が大きい傾向にあった. これは金属原子が持つd軌道に由来する配位子場の性質や金属と配位子間の電荷移動をNonbondedモデルでは評価できないためであると考えられる.
Bondedモデルでは, B3LYP/6-31G*の結果と比較するとアポ体および複合体の両者で, 結合距離の誤差は大きい傾向にあり, 結合角度の誤差は小さい傾向にあった. Bondedモデルの最適化構造は, 水分子が鉄に異常に接近しており (Figure 3a), 結合距離の誤差要因であると考えられる. このような水分子の異常な接近は, TIP3Pモデルの水分子の水素原子のLennard-Jonesパラメータが0として取り扱われることに加えて, もともと生じるべき水分子の酸素原子間の静電反発が鉄を介した1, 3-相互作用として取り扱われるため, その寄与が無視されることで生じた結果といえる[5]. したがって, 金属に水分子が配位している場合, Bondedモデルでは金属配位環境を正しく再現できないことが示唆される.

Hybridモデルでは, B3LYP/6-31G*の結果と比較すると, アポ体では結合距離および結合角度の誤差は大きい傾向にあり, 複合体では結合距離および結合角度の誤差は同程度であった. アポ体の最適化構造は, Figure 3bから確認できるように, 一つの水分子が鉄から解離しており, その結果, 結合距離および角度の誤差が大きくなったといえる. HybridモデルはNonbondedモデルと異なり, 鉄の電荷が各アミノ酸残基に分配されているため, 鉄と水分子間の相互作用が弱くなったことで, 水分子が解離したといえる (鉄のRESP電荷はHybridモデルおよびNonbondedモデルでそれぞれ, +1.1および+2.0である). 静止状態のHPPDの鉄は, 5および6配位の混合状態であることが報告[6]されており, 実際に, ヒトHPPDのアポ体において, 5配位状態のX線結晶構造 (PDB: 3ISQ, 5EC3 (中心金属はCo2+)) も報告されている. そのため, Hybridモデルで得られた5配位状態は, 生体中で存在する可能性がある. また, このような水分子の解離結果から, Hybridモデルは金属配位子の交換反応を評価できることが示唆される. 複合体に関しては, Nonbondedモデルに比べると, 結合距離および結合角度の誤差の改善傾向を確認できた. これは鉄とアミノ酸残基間の相互作用を明示的な結合として取り扱うことで, 遷移金属の配位子場や金属と配位子間の電荷移動の効果の一部を評価できたためであると考えられる.
前述のNonbondedモデルおよびHybridモデルでは, クラスターモデルにおいてHPPDの鉄配位環境を再現できる可能性があることを確認できた. そこで, アポ体のHPPD全系に対するMDを実施した. なお, MD前に行ったHPPD全系に対する構造最適化の結果, NonbondedモデルおよびHybridモデルの両者において, 鉄の配位状態は6配位であった. Hybridモデルにおいて, クラスターモデルにおける構造最適化では鉄の配位状態が5配位であったことから, HPPDの環境が鉄の配位構造の維持に影響することが示唆される.
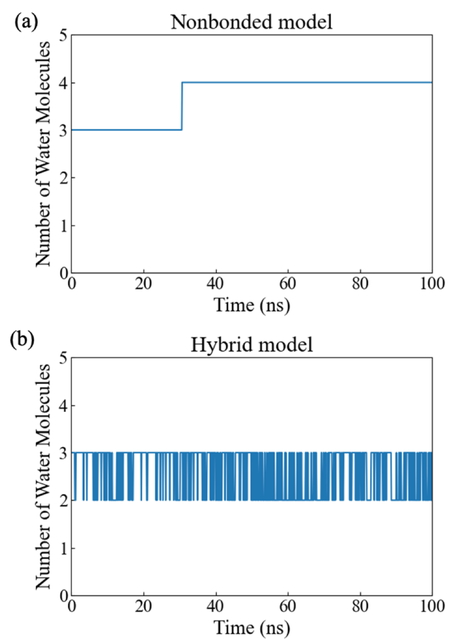
Figure 4にMD中における鉄から3.0 Å以内の水分子の個数を示す. Nonbondedモデルでは30 ns付近から水分子の個数が3から4に増加していることが確認できる. これは, 鉄と配位アミノ酸残基との間に水分子が侵入したことが原因である. そのため, Nonbondedモデルでは長時間のMDを実施した場合, 金属の配位環境を維持できないことが示唆される. Hybridモデルでは水分子の個数は2および3を行き来していることを確認できる. 上述のように, 静止状態のHPPDの鉄は5および6配位の混合状態であることが報告されていることから[6], 本MDで得られた水分子の鉄への結合・解離は妥当であると考えられる.
4 まとめ
金属含有タンパク質に対するMDでは, 金属活性中心の配位環境をHybridモデルで取り扱うことにより, 正しく再現できる可能性が高いことを確認できた. 特に, Hybridモデルは金属配位子の交換反応を評価可能であることが期待される.また, QM/MM計算[7]やより高精度なCC法などとの計算の比較[8]を実行することで, Hybridモデルの妥当性が検証されるものと考えられる. 今後は, 本パラメータを用いたMD計算によって, メソトリオンのHPPDに対する結合能や阻害能の植物種依存性[9]を検討していく方針である.
謝辞
本研究は, AMED研究課題「中分子シミュレーション」 (課題番号:JP20ae0101047h0001) による研究費支援, ならびに, 筑波大学計算科学研究センター学際共同プログラム支援のもと大型計算機Cygnusを利用して実施された.
Reference
- [1]
P. Li, B. P. Roberts, D. K. Chakravorty, K. M. Merz, Jr., J. Chem. Theory Comput., 9, 2733 (2013). doi:10.1021/ct400146w PMID:23914143
- [2]
S. C. Hoops, K. W. Anderson, K. M. Merz, Jr., J. Am. Chem. Soc., 113, 8262 (1991). doi:10.1021/ja00022a010
- [3]
A. Tomić, G. Horvat, M. Ramek, D. Agić, H. Brkić, S. Tomić, J. Chem. Inf. Model., 59, 3437 (2019). doi:10.1021/acs.jcim.9b00235 PMID:31274304
- [4]
P. Li, K. M. Merz, Jr., J. Chem. Inf. Model., 56, 599 (2016). doi:10.1021/acs.jcim.5b00674 PMID:26913476
- [5]
L. Hu, U. Ryde, J. Chem. Theory Comput., 7, 2452 (2011). doi:10.1021/ct100725a PMID:26606619
- [6]
M. L. Neidig, M. Kavana, G. R. Moran, E. I. Solomon, J. Am. Chem. Soc., 126, 4486 (2004). doi:10.1021/ja0316521 PMID:15070344
- [7]
M. Shoji, T. Murakawa, S. Nakanishi, M. Boero, Y. Shigeta, H. Hayashi, et al., Chem. Sci. (Camb.), 13, 10923 (2022). doi:10.1039/D2SC01356H
- [8]
M. Shoji, N. Watanabe, Y. Hori, K. Furuya, M. Umemura, M. Boero, et al., Astrobiology, 22, 1129 (2022). doi:10.1089/ast.2022.0011
- [9]
T. R. Hawkes, M. P. Langford, R. Viner, R. E. Blain, F. M. Callaghan, E. A. Mackay, et al., Pestic. Biochem. Physiol., 156, 9 (2019). doi:10.1016/j.pestbp.2019.01.006 PMID:31027586