Abstract
Thermodynamic properties and atomic configuration of PdRu alloy were investigated using
Wang-Landau Monte Carlo method in combination with a newly-developed universal neural
network potential. By using this new potential, excess energy of PdRu alloy was
calculated. It is found that PdRu alloy in FCC lattice is unstable in the full range of
alloy composition. This agrees with previous study based on density functional theory. The
combined method was able to determine the configurational density of states, from which
thermodynamic properties of the alloy were derived. It is found that when temperature
increases, the excess free energy of the alloy is reduced, increasing the possibility of
alloy mixing. Depending on the composition, transition peaks appear at finite temperatures
where there are changes of preferable atomic arrangement due to the effect of temperature
via the configurational entropy. In addition, the analyses on short-range order parameter
and bond fraction show that PdRu alloy prefers to be in segregated form, where Pd and Ru
are immiscible at low temperature, consistent with the experimental observations. The
random mixing of Pd and Ru atoms in the form of solid-solution can occur at high
temperature.
1 Introduction
The discovery of novel alloy and composite materials with good performance for specific
applications has been a hot topic in the field of materials science. For instance, the
materials discovery is being applied to the research on the improvement of catalytic
performance and long-term stability of electrodes in fuel cells [1, 2] or the research on the
improvement of the cathode life cycle under charging and discharging processes in Li-ion
batteries by mixing different metal elements with different compositions [3]. Traditionally, materials discovery often consumes a
vast amount of time and resources in experiments and computational simulations because of
the limitations of experimental conditions or theoretical foundations. Thus, it is crucial
to approach this task in a new way. In recent years, due to the availability of large
experimental and simulation datasets in combination with advanced algorithms and computer
resources, materials discovery using data-driven and machine learning approaches has been
receiving increasing attention as well as achievements in both time efficiency and
prediction accuracy. One of the approaches in this new field is to use neural network
potentials (NNP) [4], which model the interactions of
atoms by mimicking the neural networks in the human brain. These kinds of potentials are
expected to retain the accuracy up to the quantum mechanical calculation level and speed up
to the force field calculation level. In addition, due to the rapid development of advanced
computational techniques, such as the multicanonical Monte Carlo method [5], a method for predicting the stability of alloys can be
implemented.
2 Methodology
2.1 Wang-Landau sampling
Thermodynamic properties of alloys are calculated based on the multicanonical ensemble
method [5, 6].
Here, instead of sampling the probability distribution at a fixed temperature in
configurational space as usually carried out in conventional Metropolis Monte Carlo (MC)
methods, energy space is explored to estimate the configurational density of states (DOS),
g(E). This is a central quantity in the Wang-Landau
(WL) sampling technique [7, 8]. Once this is known, the partition function can be calculated from
[9]:
|
Z
=
∑
E
g
(
E
)
e
−
E
/
k
b
T
| (1) |
From the partition function above, other thermodynamic quantities of a given system such
as free energy (F), internal energy (U), and specific
heat (Cv) can be derived:
|
U
=
1
Z
∑
E
E
g
(
E
)
e
−
E
/
k
b
T
| (3) |
|
C
v
=
1
k
b
T
2
(
⟨
E
2
−
U
2
⟩
)
| (4) |
In the WL sampling method, the probability of transition from state A to B is evaluated
from:
|
p
(
E
A
→
E
B
)
=
min
(
1
,
g
(
E
A
)
/
g
(
E
B
)
)
| (5) |
Trial move is performed by swapping two arbitrarily atoms of dissimilar types in the
alloy compound. The sampling starts with an initial guess of
g(E) (=1) and zero-component energy histogram,
H(E). Every time when a trial move is accepted,
g(E) and H(E) of new
state are updated. Otherwise, those of old state will be modified. The updates are done by
multiplying g(E) with a modification factor,
f, and adding 1 to H(E). The
criterion for stopping the updates for H(E) is if any of
its value is larger than 0.8 multiplied by its average. At this point,
H(E) is considered flat with a flatness criterion of
0.8. When this condition is met, f will be reduced to
f1/2 and H(E) is reset to
0. The initial value of f was set as the Napier's constant (
e
=
2.71828
…
) and the criterion for stopping the sampling is when
f is less than
e
x
p
(
10
−
8
)
≈
1
+
10
−
8
. This means that the difference between the configurational
DOS of the two last iterations is about 10−8, and hence the final
configurational DOS is considered to be the true DOS. The flatness criterion for
H(E) was chosen based on the suggestions by Wang and
Landau [7]. In addition to it, further study showed
that while the results of Wang-Landau simulations are excellent with a flatness criterion
of 0.95, the results for the case of 0.8 are quite adequate [10]. The choice of the final modification factor is based on the fact
that when the transition peak of the specific heat is considered, the ideal limit for
f should be exp(10−4) or smaller [11].
2.2 Energy calculations
For each trial move in the WL sampling, total energy of the given alloy compound is
calculated. This is the most time-consuming part in WL method as well as other
conventional MC techniques. To accelerate the energy calculations, a recently developed
universal NNP, namely Preferred
Potential (PFP version 2.0.0) [12], is used due to its calculation speed, versatility, and wide-range of
support for many elements. There are several calculation modes in PFP. In the case of PdRu
alloy, the mode for crystal systems with Hubbard and dispersion corrections
(CRYSTAL_PLUS_D3) is selected.
To examine the accuracy of this potential, excess energy of PdRu alloy is calculated and
compared with the results from density functional theory (DFT). Here, the excess energy is
estimated by:
|
E
e
x
=
(
E
P
d
R
u
−
N
P
d
ϵ
P
d
F
C
C
−
N
R
u
ϵ
R
u
H
C
P
)
/
N
| (6) |
where
ϵ
P
d
F
C
C
and
ϵ
R
u
H
C
P
are the total energies per atom of FCC Pd and HCP Ru,
respectively and
E
P
d
R
u
is the total energy of PdRu alloy supercell containing
N
P
d
Pd and
N
R
u
Ru atoms (
N
P
d
+
N
R
u
=
N
).
In this work, a 4 × 4 × 4 supercell of FCC PdxRu1-x (x = 0.25,
0.50, 0.75) alloy containing 256 atoms is used for WL sampling. To speed up the evaluation
of energy, all atomic positions and cell volume/shape were kept fixed with pre-optimized
lattice constants, which are 3.797 Å, 3.824 Å, and 3.858 Å for
Pd64Ru192, Pd128Ru128, and
Pd192Ru64, respectively. While, for the comparison of excess
energies by PFP and DFT calculations, a 2 × 2 × 2 supercell was considered. DFT
calculations were conducted using VASP [13, 14], where the exchange-correlation functionals based
on the Perdew-Burke-Ernzerhof generalized gradient approximation [15] was used, the electron-core interactions are described by the
projector-augmented wave method [16, 17]. Optimization criterion for atomic forces is below
0.02 Å/eV when applicable. For the consistency with PFP calculations, dispersion
correction with Becke-Johnson damping (DFT-D3 (BJ)) [18] was introduced. Furthermore, to examine the performance of PFP, calculation
time was monitored. Using the 2 × 2 × 2 supercell as a reference, we found that the
optimization time for the Pd128Ru128 is about 2.1 seconds, while for
DFT calculation running on a 32-core Intel Xeon CPU, it took about 9.2 hours. For fixed
total energy calculations, calculation time are 0.1 seconds and 1.1 hours for PFP and DFT,
respectively.
2.3 Short-range order parameters
To evaluate the atomic ordering in PdRu alloy from WL simulation, the Warren-Cowley
short-range order (SRO) parameter [19, 20] was used. In binary alloy, the SRO parameter of
dissimilar types is expressed as:
|
α
A
B
=
1
−
P
A
B
/
c
B
| (7) |
Here, PAB and cB represent the
probability of finding the B atom surrounding the A atom
and the overall composition of B atom in the compound, respectively. When
αAB is close to zero, it means that two elements are
randomly mixed.
3 Results and Discussion
3.1 Excess energy
In Figures 1 (a) and (b), the excess energies
calculated from DFT and PFP are compared. Here, 30 random configurations with different
alloy compositions and 3 ordered structures with respect to L12-type and
L10-type orderings are chosen. We can see that the consistency of excess
energies is better for the ordered structures than the random ones. The coefficients of
determinant (R2) from fixed and relaxed calculations are 0.660
and 0.674, respectively. These values are moderate in the PFP version 2.0.0 but further
checks for latest version (3.0.0) showed that PFP excess energies are comparable to DFT
values, 0.926 and 0.956 for fixed and relaxed calculations, respectively. The difference
of R2 between the fixed and relaxed calculations are very
small, for both PFP versions. This means that energy calculations using PFP are well
consistent with DFT. In addition, the compositional dependence of excess energy was also
examined for FCC PdRu alloy and is shown in Figure 1
(c). Here, calculations using PFP were carried out by alloying Ru in FCC Pd
lattice at different concentrations in both the randomly mixed and segregated atomic
arrangements. For each value of Ru concentration in the randomly-mixed case, 2000 atomic
configurations were considered. We can see that PdRu alloy is unstable in the whole range
of Ru composition. For the randomly-mixed arrangement, the maximum mean value of excess
energy is about 0.135 eV per atom with the maximum error bar (standard deviation) of about
0.008 eV at Ru concentration of 0.4-0.5. The segregated arrangement of atoms seems to be
more preferable than the randomly-mixed cases. In the Pd- or Ru-rich conditions, the alloy
tends to be more stable. The results calculated by PFP in this work is in good agreement
with previous work using DFT [21].

The configurational DOS for 3 PdRu alloy compositions (Pd64Ru192,
Pd128Ru128, and Pd192Ru64) were estimated
from WL sampling and depicted in Figure 2 (a).
The range of excess energy is smaller than that calculated by regression equation [21]. This is because the energy range of each alloy was
pre-estimated from two randomly-mixed atomic configurations. For each alloy composition,
the two configurations above are the most stable and unstable ones among more than 10
million scanned structures. As shown in previous sub-section, the excess energy of
randomly-mixed arrangements is larger than that of segregated one. Hence, the excess
energy range is smaller in this work. From the configurational DOS, thermodynamic
quantities were calculated using thermodynamic equations together with the partition
function in equation 1. In Figures 2 (b) and
(c), the excess free energy and specific heat are presented. It is noted that in
the WL simulation, only contribution from atomic arrangement (or configurational
contribution) was included in calculation of thermodynamic properties. Vibrational
contribution was not considered in this current work. In principle, the two phenomena
above are system-dependent. However, in the case of PdRu alloy, one expects that the
former is more pronounced than the latter (i.e. the latter offsets between different
configurations) [22]. We can see that when
temperature increases, the excess free energies in the three compounds are decreased due
to the contribution of configurational entropy. The change is more dramatical for
Pd128Ru128. This might be the fact that
Pd128Ru128 with Ru concentration of 0.5 is very much unstable as
mentioned in previous sub-section. When temperature increases, the configurational entropy
works to stabilize the system more significantly. Although the attenuation of the excess
free energy with increase in temperature is obvious, they are still positive. This means
that the PdRu compound is unstable than the monometallic systems.
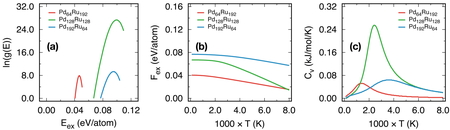
From the configurational specific heat, we can see that peaks appear in all
C
v
curves. These peaks indicate the change of preferable
atomic arrangement from one to another due to the effect of temperature via the
configurational entropy. Obviously, with different composition, the transition
temperatures for the three compounds are different. The lowest is for
Pd64Ru192 (about 1300K) and the highest is for
Pd192Ru64 (3400 K). In the case of
Pd128Ru128, the transition temperature is about 2400 K. Comparing
these values with the results of previous work in which the total energy was calculated
using supervised learning [21], it was found that
the transition temperatures for the three compounds in this study are higher. One of the
reasons for this discrepancy is that the total energy calculation in this work was applied
to fixed supercell and atomic positions, that makes the excess energy ranges narrower for
all compounds as we can see from configurational DOS in Figure 2 (a). The narrowness of energy range might also be due to the choice of
the initial configuration in WL simulation. Further analysis has shown that the choice of
energy range in WL sampling is critical to produce correct solution for the thermodynamic
properties of alloy compounds. This is illustrated in Figure 3. Assuming that some segments are cut out from the full range
configuration DOS (Figure 3 (a)) and the
specific heat is calculated for each segment. We can see in Figures 3 (a), (b), and (c) that the shapes of newly-generated
specific heat curves and the positions of transition temperatures are changed. The change
is less for the first segment where the WL sampling has visited the most stable
configuration. However, the other two segments, two transition peaks disappear. Even for
the third segment, the transition peak shifts far away from the specific heat calculated
from the full range DOS to higher temperature. Hence, the value of transition temperature
is strongly dependent on how the energy segment is taken in the WL sampling. To improve
this, a procedure to pre-determine approximately the stable and unstable configurations at
0K should be deployed before the sampling process.

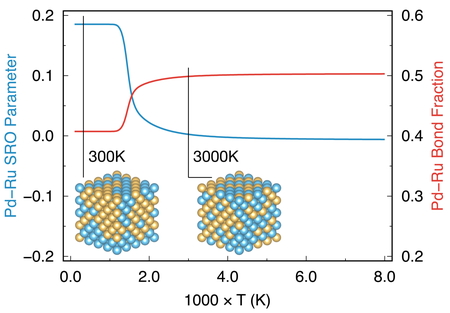
The SRO parameter as well as the bond fraction in alloy compounds are thermodynamic
observables and can be estimated from WL simulation via the 2-dimensional configurational
DOS, g(E,σ). Here, the second variable
in g(E,σ) is the thermodynamic
observable. The results of Warren-Cowley SRO parameter and Pd-Ru bond fraction as
functions of temperature in Pd128Ru128 alloy is shown in Figure 4. These two quantities are in principle
interchangeable as shown in equation 7 and lead to similar discussion on atomic ordering
in the alloy. We can see that at low temperature, the Pd-Ru bond fraction and SRO
parameter are about 0.4 and 0.2, respectively. These correspond to the segregation of Pd
and Ru atoms in the alloy. The result is consistent with the experimental works which
stated that at room temperature, there is no combination of Pd and Ru in the form of solid
solution in bulk alloy [23]. This happens only when
they are in the form of nanoparticles [24]. When
temperature overcomes the transition point (around 1200 K, corresponding to the highest
peak in the specific heat curve in Figure 3
(b)), the bond fraction and the SRO parameter are drastically changing. At very
high temperature, the values are 0.5 and 0.0 for the bond fraction and the SRO parameter,
respectively. These indicate a random mixture state of Pd and Ru atoms. In this case, the
probability of finding A surrounding B is large and vice versa.
4 Conclusion
In summary, we have successfully combined current advanced techniques, namely the
Wang-Landau sampling and universal neural network potential, to study the thermodynamic
properties and atomic arrangement in metallic alloy compounds. The method has been applied
to study the stability of PdRu alloy. The novel neural network potential was proven to
improve significantly the calculation speed as compared to the state-of-art DFT method. At
the same time, it reproduced well the composition-dependent excess energy of PdRu alloy when
compared to other study using DFT and supervised learning model. Furthermore, the combined
method was shown to be a good way to investigate the contribution of configurational entropy
to the thermodynamic properties and atomic ordering of alloys. It can reproduce well the
atomic mixing phenomena in PdRu as compared to previous works. Obviously, this method can be
applied not only to the PdRu alloy but also to the study any kind of alloy systems, due to
the "universal" feature of the neural network potential. However, it is very important to
choose the energy range in WL sampling. This can strongly govern the accuracy description of
the thermodynamic properties of alloy systems.
Acknowledgement
This work was supported by the JST CREST (Grant No. JPMJCR21B3) and JSPS KAKENHI (Grant No.
20H05623). The calculations were partly conducted using the Supercomputing system resources
(MASAMUNE-IMR) from the Center for Computational Materials Science of the Institute for
Materials Research, Tohoku University.
References
- [1]C. Li, K. Wang, and D.
Xie, Surf. Interfaces, 28, 101594(1–11) (2022).
- [2]M. Du, X. Li, H. Pang, and
Q. Xu, EnergyChem, 5, 100083(1-37) (2023).
- [3]J. U. Choi, N. Voronina,
Y.-K. Sun, and S.-T. Myung, Adv. Energy Mater., 10, 2002027(1–31) (2020).
- [4] J. Behler, Chem. Rev., 121,
10037 (2021). doi:10.1021/acs.chemrev.0c00868 PMID:33779150
- [5] B. A. Berg, T. Neuhaus,
Phys. Lett. B, 267, 249 (1991). doi:10.1016/0370-2693(91)91256-U
- [6] B. A. Berg, T. Neuhaus,
Phys. Rev. Lett., 68, 9 (1992). doi:10.1103/PhysRevLett.68.9
PMID:10045099
- [7] F. Wang, D. P. Landau, Phys.
Rev. Lett., 86, 2050 (2001). doi:10.1103/PhysRevLett.86.2050
PMID:11289852
- [8] D. P. Landau, F. Wang,
Comput. Phys. Commun., 147, 674 (2002). doi:10.1016/S0010-4655(02)00374-0
- [9] D. P. Landau, S.-H. Tsai,
and M. Exler, Am. J. Phys., 72, 1294–1302 (2004).
- [10]P. H. Nguyen, E. Mittag,
A. E. Torda, and G. Stock, J. Chem. Phys., 124, 154107(1–7) (2006).
- [11]A. A. Caparica, Phys.
Rev. E., 89, 043301(1–7) (2014).
- [12]S. Takamoto, C.
Shinagawa, D. Motoki, K. Nakago, W. Li, I. Kurata, T. Watanabe, Y. Yayama, H. Iriguchi, Y.
Asano, T. Onodera, T. Ishii, T. Kudo, H. Ono, R. Sawada, R. Ishitani, M. Ong, T.
Yamaguchi, T. Kataoka, A. Hayashi, N. Charoenphakdee, and T. Ibuka, Nat. Commun., 13,
2991(1–11) (2022).
- [13] G. Kresse, J. Furthmüller,
Phys. Rev. B Condens. Matter, 54, 11169 (1996). doi:10.1103/PhysRevB.54.11169
PMID:9984901
- [14] G. Kresse, J. Furthmüller,
Comput. Mater. Sci., 6, 15 (1996). doi:10.1016/0927-0256(96)00008-0
- [15] J. P. Perdew, K. Burke, M.
Ernzerhof, Phys. Rev. Lett., 77, 3865 (1996). doi:10.1103/PhysRevLett.77.3865
PMID:10062328
- [16] P. E. Blöchl, Phys. Rev. B
Condens. Matter, 50, 17953 (1994). doi:10.1103/PhysRevB.50.17953
PMID:9976227
- [17] G. Kresse, D. Joubert, Phys.
Rev. B Condens. Matter, 59, 1758 (1999). doi:10.1103/PhysRevB.59.1758
- [18]A. D. Becke and E. R.
Johnson, J. Chem. Phys., 122, 154104(1–5) (2005).
- [19] J. M. Cowley, Phys. Rev.,
77, 669 (1950). doi:10.1103/PhysRev.77.669
- [20] J. M. Cowley, Phys. Rev.,
138, 5A, A1384 (1965). doi:10.1103/PhysRev.138.A1384
- [21] Y. Nanba, M. Koyama, Phys.
Chem. Chem. Phys., 24, 15452 (2022). doi:10.1039/D2CP01848A PMID:35712830
- [22] T. Ishimoto, M. Koyama,
arXiv:2007.06137v1 [cond-mat.mtrl-sci], https://doi.org/ /arXiv.2007.06137
(2020).doi:10.48550
- [23] S. N. Tripathi, S. R.
Bharadwaj, M. S. Chandrasekharaiah, J. Phase Equilibria, 17, 362 (1996).
doi:10.1007/BF02665565
- [24] K. Kusada, H. Kobayashi, R.
Ikeda, Y. Kubota, M. Takata, S. Toh, et al., J. Am. Chem. Soc., 136, 1864 (2014).
doi:10.1021/ja409464g PMID:24455969