実践報告
立命館大学におけるレギュラトリーサイエンス教育の事例
―医薬品の模擬承認審査
2021 年 5 巻 論文ID: 2020-048
詳細
2021 年 5 巻 論文ID: 2020-048
レギュラトリーサイエンス教育の一貫として,2018年度から臨床試験概論(8コマ)の中で医薬品の模擬承認審査を実施している.同年度は事前に審査対象品目名を開示し,審査側と申請者側に分けて実施したところ,審査結果等が審査報告書と同じ,主要な論点が絞り切れず発表が冗長,審査側・申請者側の役割が生かしきれない等の問題があった.このため,2019年度は実施方法を改善し,品目名を伏せた既承認の抗インフルエンザウイルス剤2品目を対象とし,それらの基本情報や臨床試験成績概略等をPMDA医療用医薬品情報検索サイトから収集し,学生(4回生70名)に提示した.また,論理的思考力の向上及び議論の活発化のため,学生を課題毎に承認可・不可グループに分けた.その結果,活発な議論が行われ,グループ毎に纏めた審査結果等の発表も論点が絞られすべて時間内に終了した.さらに実施方法を改善し,今後も模擬承認審査による教育を継続したい.
The regulatory science education at Ritsumeikan University includes approving mock drug reviews in their Introduction to Clinical Studies course (a total of 8 classes). In 2018, the first year of this course, the students divided into reviewer and applicant groups for discussions with the names of the review products disclosed beforehand. Some issues that resulted from this activity were: (1) the results of the drug review were similar to the review reports by PMDA, (2) the presentations became redundant because they did not narrow down the main points, and (3) the different roles of reviewer and applicant were not fully utilized. To improve this learning method in 2019, 70 senior students were provided with the basic drug information and summaries of clinical studies for two approved anti-influenza viral agents. The drug names were not disclosed, and the information was obtained from the PMDA’s ethical pharmaceutical information search website. Students were divided into two groups to approve or deny each drug to improve logical thinking ability and to stimulate discussion. The outcome was that they debated actively, and each group successfully presented the collated review results within the time limits. Our objective is to continue improving on this learning method with these mock drug reviews.
「レギュラトリーサイエンス」(Regulatory Science:以下,「RS」)の対象分野は広範であるため,その定義には様々なものがあるが趣旨は同じである.医療分野の研究開発等に関しては,健康・医療戦略推進法(平成26年5月)で「(医療分野の研究開発の成果の実用化に際し,その)品質,有効性及び安全性を科学的知見に基づき適正かつ迅速に予測,評価及び判断することに関する科学」とされ,また,「国は,RSの振興に必要な体制の整備,人材の確保,養成及び資質の向上その他の施策を講ずる」旨が明記された1).さらに,薬学教育モデル・コアカリキュラム(平成25年度改訂版)2) には,「RSの必要性と意義について説明できる.」旨がB「薬学と社会」の到達目標(specific behavioral objective: SBO)のひとつとされている.薬学の分野におけるRSは,医薬品等の品質・有効性・安全性確保のための科学的方策の研究や試験法の開発,実際の規制のためのデータの作成と評価などであり3),薬学教育で履修する幅広い専門領域が関わるとされている4).
立命館大学薬学部では,様々な科目を通じてRS教育を行っているが,2018年4月から『レギュラトリーサイエンス研究室』を新たに立ち上げ,RS研究を通じた教育にも力を注いでいる.このうち臨床試験概論(選択科目)では,2017年度までは6回生数名を対象とした15回授業科目で臨床試験実施計画書や症例報告書の作成を行っていた.2018年度からは4回生を対象とした8回授業となったため,臨床試験成績の評価や臨床試験の実施に必要な検討項目の理解,リスクとベネフィットのバランスの判断及びRSの理解に役立ち,また臨床試験概論の目標の達成及び論理的な思考力の向上にも資すると考え,学生による医薬品の模擬承認審査(以下,「模擬審査」)を実施することにした.
2018年度は,履修生(4回生)を,審査対象品目ごとに申請者側と審査側のグループに分けて模擬審査を行った.審査対象は既承認の新有効成分含有医薬品4品目とし,それらに関する情報(審査報告書,申請資料概要,添付文書等)は,学生が(独)医薬品医療機器総合機構(以下,「PMDA」)ホームページの医療用医薬品情報検索サイト5) から収集することとしたため,予め品目名が開示された状態であった.模擬審査の結果等の発表は,承認申請された品目について,PMDAにおける審査の手順のうち最初に実施される「初回面談」(審査側から提示される品目の主要な論点について申請者と議論する面談)と類似の方法で実施した.その結果,模擬審査の結果等が審査報告書と同様で学生の考えが表れてこなかったこと,主要な論点が絞りきれず発表が冗長になったこと,審査側・申請者側の役割が生かし切れていなかった等の問題がみられた.このため,2019年度は実施方法を改善し,対象の医薬品の品目名を伏せ,事前に承認の可否別にグループ分けしたうえで模擬審査を実施した.なお,品目の基本情報及び臨床試験成績概略等は,教員が提供した.
現時点で模擬審査によりRS教育を実施している大学・大学院はあるが6),具体的な実践例の報告はないことから本事例を紹介する.
2019年度模擬審査の実施手順を表1に示した.模擬審査では医薬品の承認申請書に添付されている臨床試験成績の評価ができるように,臨床試験概論における一連の講義に加えて,「臨床試験成績に関する審査の観点」として,①臨床データパッケージ,②疾患概念,③品目の臨床上の位置づけ,④対象患者の妥当性,⑤評価項目の妥当性,⑥有効性が認められているか,⑦安全性に問題がないか,⑦用法・用量,効能・効果の妥当性,⑧医薬品リスク管理計画(Risk Management Plan: RMP)は適切か,製造販売後調査の内容,⑨添付文書,⑩薬剤のパッケージ,⑪医療従事者・患者向け資材等について説明した7,8).その上で,グループ学習,成果発表,個人評価としてのレポート提出を行った.
| 1.臨床試験成績に関する審査の観点を講義 |
| ①臨床データパッケージ,②疾患概念,③品目の臨床上の位置づけ,④対象患者の妥当性,⑤評価項目の妥当性,⑥有効性が認められているか,⑦安全性に問題がないか,⑦用法・用量,効能・効果の妥当性,⑧医薬品リスク管理計画(Risk Management Plan: RMP)は適切か,製造販売後調査の内容,⑨添付文書,⑩薬剤のパッケージ,⑪医療従事者・患者向け資材等 |
| 2.医薬品候補の販売名および一般名を各グループが付すことで,名称ルールの一端を学ぶ |
| 3.模擬審査の実施(グループ学習,発表)により,臨床試験実施に際して必要な項目,試験成績の評価について理解する. |
| 1)提示した情報:医薬品候補の基本情報(作用機序,剤形・含量,効能・効果,用法・用量,申請年月)に加えて,臨床試験一覧,臨床試験デザインの概略(被験者,評価項目,症例数,解析方法等),臨床試験成績の概略(第II相,第III相),類薬の情報 |
| 2)医薬品候補(候補A,候補B)それぞれについて,承認可・承認不可グループに分け,課題検討.なお,評価項目は妥当とする. |
| 3)検討内容:①臨床上の位置づけ,②有効性は認められているか,③安全性は忍容可能か,④耐性について,⑤臨床試験成績からみて効能・効果は適切か,⑥臨床試験成績からみて用法・用量は適切か,⑦承認可・不可の判断根拠.不足している情報の有無 |
| 4)グループ学習成果の発表(プレゼンテーション各13分,質疑応答7分),解説 |
| 質疑応答に際しては,承認可グループと不可グループ相互で質問・反論を行う. |
| 5)発表の評価 |
| 各グループに対する評価は,以下の観点から行った(学生には発表前に説明). |
| ①論点が明確か(0~5点),②論旨に矛盾がないか(0~5点),③問題点に対する対応策を考えているか(0~3点),④説得力のある説明か(0~3点),⑤分かりやすいか(0~2点),⑥印象に残ったか(0~2点),⑦話す早さは適切か(0~2点),⑧時間内に終わったか(0~2点) |
| 4.個人の評価:レポート提出 |
| 発表で使用したスライドに加えて,以下の項目について論述. |
| 1)当日の質疑応答の内容,自らの考え及び考察 |
| 2)検討の過程で問題点として掲げられていたこと,時間の都合で説明できなかったこと |
| 3)承認の可否に関する個人の意見(可・不可,その根拠・理由) |
| 4)模擬審査に関する感想 |
模擬審査の対象品目は,学生が興味を持ちやすく,他の疾患に比べ理解しやすい抗インフルエンザウイルス剤とし,既承認品目から選択した.品目名は伏せ「医薬品候補A,B」として提示した.2018年度の模擬審査において審査側・申請者側の役割が生かし切れなかったことを踏まえ,また,思考力(論理的思考力,瞬時に考え判断する能力,批判的思考力),発信力,傾聴力のスキルを獲得するために,臨床試験概論の履修者(4回生)70名を4グループに分け,医薬品候補A,Bに対してそれぞれ承認可・不可グループに割り振った.なお,各品目の臨床試験(治験)における評価項目は妥当を前提とした.各品目の申請時の基本情報(作用機序,剤形・含量,効能・効果,用法・用量,申請年月),臨床試験一覧,臨床試験デザインの概略(被験者,評価項目,症例数,解析方法等),臨床試験成績の概略(第II相,第III相),類薬の情報をPMDAの医療用医薬品情報検索サイト5) から収集し,品目名(販売名,一般名)を伏せて提示した.そして,これらの情報から,①臨床上の位置づけ,②有効性は認められているか,③安全性は忍容可能か,④耐性について,⑤臨床試験成績からみて効能・効果は適切か,⑥臨床試験成績からみて用法・用量は適切か,⑦承認可・不可の判断根拠,不足している情報について検討させた.
最後に,グループ学習の成果発表(プレゼンテーション13分,質疑応答7分)を行った.なお,評価の観点を表1に示した.発表が終了した後,医薬品候補A,Bの品目名を明かした.
2.レポート以下①~④について論述したレポートを提出させた.①当日の質疑応答の内容,自らの考え及び考察,②検討過程で問題点として掲げられていたことや時間の都合で説明できなかったこと,③承認の可否に関する個人の見解とその根拠・理由,④模擬審査に関する感想.
医薬品候補A(ペラミビル水和物)9) の承認可・不可グループによる発表内容の概略を表2(a)に示した.
| (a)医薬品候補A:ペラミビル水和物 | ||
|---|---|---|
| 承認可グループ | 承認不可グループ | |
| 臨床上の位置づけ | ✓他剤無効な症例に対する「取って置き」の薬ではなく,経口や吸入のNA阻害薬と同様,第1選択に位置する | ✓NA阻害薬の選択肢が増える ✓経口薬や吸入薬で効かなかった人の第2選択となる |
| 有効性 | ✓プラセボに比べて罹病期間が短縮(第II相) ✓オセルタミビルと有意差がない(第III相) ✓A型:オセルタミビルと同等の有効性,B型:300 mgで有効性に期待 |
✓第II相:B型のプラセボ群の患者数が0では比べられず,有効性は認めることができない |
| 安全性 | ✓投薬中止に至った有害事象,死亡例はなかった ✓重篤な副作用:本剤300 mg群(3日投与)に細菌性肺炎1件,600 mg群(2日投与)に肺炎1件,因果関係は否定 ✓投与中止するほどの有害事象は認められていない →安全に実用化できる可能性が高い |
本治験薬との因果関係が認められた有害事象 ✓本剤300 mg投与による薬疹(1件) ✓本剤600 mg投与により上腹部痛,関節痛,薬疹,発疹及び蕁麻疹(各1件) |
| 効能・効果 | A型またはB型インフルインザウイルス感染症 ✓A型及びB型への有効性があると判断 ✓予防まで効果があるのか不明 ✓作用機序が新規でないため,副作用や効能・効果が予測しやすい |
✓A型:オセルタミビル群と比較して有意差なし→有効 ✓B型:被験者数がA型(約320例)に比べて少ない(本剤300 mg:21例,600 mg:26例,オセルタミビル:23例) →有効であると判断し,認証するには,検証試験が不十分 |
| 用法・用量 | ✓重症患者には反復投与とあるが,反復投与時の副作用発生リスクが高かったため,反復投与が可能なのか吟味する必要がある(用法・用量に注意する) ✓静注を基本とするため,嚥下困難な患者に対して特に期待できる. |
第II相,第III相における用法・用量から,申請時用法・用量で適切と考える |
A型:A型インフルエンザウイルス,B型:B型インフルエンザウイルス
| (b)医薬品候補B:パロキサビル マルボキシル | ||
|---|---|---|
| 承認可グループ | 承認不可グループ | |
| 有効性 | ✓成人患者対象国内第II相臨床試験: ・本薬群の罹患期間がプラセボ群より短い →A型,B型に効果あり ・B型:投与量を増やしても罹患期間の短縮が認められない →B型よりA型の方に効果が出やすい ✓12歳以上の患者対象第III相臨床試験:特にB型で罹患期間が長い |
✓成人患者対象国内第II相:プラセボ群と比べて有意に優れていた ✓12歳以上の患者対象第III相:プラセボ群と比べて罹病期間が有意に優れ(全体,A型),20歳以上65歳未満で有意差なし ✓20歳以上のB型に対する症例数が少なく,B型に効果があるというのは困難 ✓12歳未満の小児患者対象第III相:プラセボ群がないため比較できない 有効性が示されておらず,症例数を増やした第III相試験が必要 |
| 民族差について | ✓最高血中濃度はアジア人が非アジア人より高値 ✓アジア人では中央値がプラセボ及び白人に比べて小さい人種差あり,非アジア人よりアジア人において効果が得やすい →A型,B型に効果あり.民族差があるため,理解した上で使用する必要あり. |
✓第I相(健康成人)及び第III相(患者):Cmax,AUCは外国人より日本人で約2倍高く,体重は外国人が重かった.体重の違いが薬物動態パラメータに影響しており,民族差は認められる. ✓12歳以上の患者対象第III相:外国人は日本人より約2倍罹病期間が長く,有効性に民族差あり 体重による用量調節がなされているが,効果に民族差が認められるため,承認は否定的 |
| 耐性について | ― | ✓12歳未満の患児で,本薬投与後のアミノ酸変異ウイルスの出現率が高い →12歳未満で特に耐性株が出現しやすい →有効性が低下 →用法・用量や注意喚起等を見直すべき ✓12歳以上の患者対象第III相:本薬投与4日後からアミノ酸変異を有する患者ではウイルス力価がプラセボ群より高かった |
| 安全性 | ✓有害事象発現率:本薬群とプラセボ群間に大きな違いなし ✓重篤な有害事象:ウイルス性髄膜炎・嵌頓鼠径ヘルニア(12歳以上の患者対象第III相で各1件:回復,因果関係なし) ・死亡例なし.投与中止に至った有害事象発現率は,プラセボ群と大きな違いなし(本剤群:気管支炎2例,肺炎1例.因果関係なし) ✓成人患者対象国内第II相/12歳以上の患者対象第III相併合:肝機能障害関連の有害事象発現率が2%以上の群はなく,プラセボ群と大差なし ✓12歳未満の小児患者対象第III相:AST及びALT増加(因果関係あり)各1例(0.9%) →肝機能障害:製造販売後も情報収集し,医療現場に情報提供 |
成人患者対象国内第II相/12歳以上の患者対象第III相 ✓重篤な有害事象:ウイルス性髄膜炎,嵌頓鼠径ヘルニア(第III相臨床試験各1例) ✓死亡例はなくプラセボと有意な差は認められないため,12歳以上の患者では安全 12歳未満の小児患者対象第III相 ✓プラセボ群と比較していない ✓先の2試験に比べ有害事象の割合が大きく,10%を超える有害事象がある →12歳未満の使用において安全であるとは言えない ✓主な有害事象:感染症及び寄生虫症(13.1%),胃腸障害(15.0%) ✓肝機能障害:AST及びALTの増加(因果関係あり)1例 →注意喚起が必要 |
| 効能・効果 | A型及びB型の両方に効果があるが,特にA型の方に効果が出やすい | 12歳以上の患者対象第III相:罹病期間が,A型ではプラセボ群よりも本薬群が短く,B型では本薬群が長かった →B型への治療効果が示されておらず,効能・効果はA型のみに限定 |
| 用法・用量 | ✓成人患者を対象とした第II相試験から,投与量40 mgが効果的 | ✓12歳未満の小児のうち,体重40 kg以上は8例,体重5 kg以上10 kg未満は安全性評価例数2例と少ない. →例数を重ねてから,適切な用法・用量を判断すべき |
A型:A型インフルエンザウイルス,B型:B型インフルエンザウイルス
臨床上の位置づけは,承認可・不可両グループともにノイラミニダーゼ阻害剤の選択肢が増えるとしていたが,承認可グループは第1選択薬,承認不可グループは第2選択薬とした.
有効性については,いずれのグループもA型インフルエンザウイルス感染(以下,「A型」)に対する有効性は認められたと評価した.一方,B型インフルエンザウイルス感染(以下,「B型」)に対しては,承認可グループが300 mgで有効性が期待できると判断し,承認不可グループはA型に比べて被験者数が少なく有効と判断するには検証試験が不十分と判断した.
安全性については,両グループともに問題ないと評価した.
効能・効果について,承認可グループは予防効果までは不明であるが認められると判断し,承認不可グループはA型には有効だがB型には不十分と判断した.
2)医薬品候補B(パロキサビル マルボキシル)について医薬品候補B(パロキサビル マルボキシル)10) の承認可・不可グループによる発表内容の概略を表2(b)に示した.
有効性については,いずれのグループもA型に対する有効性は認められたと評価した.一方,B型に対しては,承認可グループがB型よりA型に対して効果が出やすいとしており,承認不可グループもB型に効果があるとは言い難く,有意差が認められない年齢層があることに言及していた.また,承認不可グループは民族差があるため承認に否定的とし,承認可グループは理解した上で使用する必要があるとした.耐性については,承認不可グループのみが評価を行い,12歳未満で特に耐性株が出現しやすく有効性が低下すること,用法・用量や注意喚起を見直すべきとした.
安全性については,両グループともに肝機能障害に関する注意喚起が必要であるとした.
効能・効果については,承認可グループはA型及びB型ともに効果が認められるが,特にA型に対して効果が認められやすいとした.一方,承認不可グループは,B型に対する治療効果が示されていないとして,効能・効果は「A型インフルエンザウイルス感染症」に限定すべきとした.
2.承認の可否に関する個人の意見承認可・不可のグループ別の個人の意見のまとめを図1に示した.
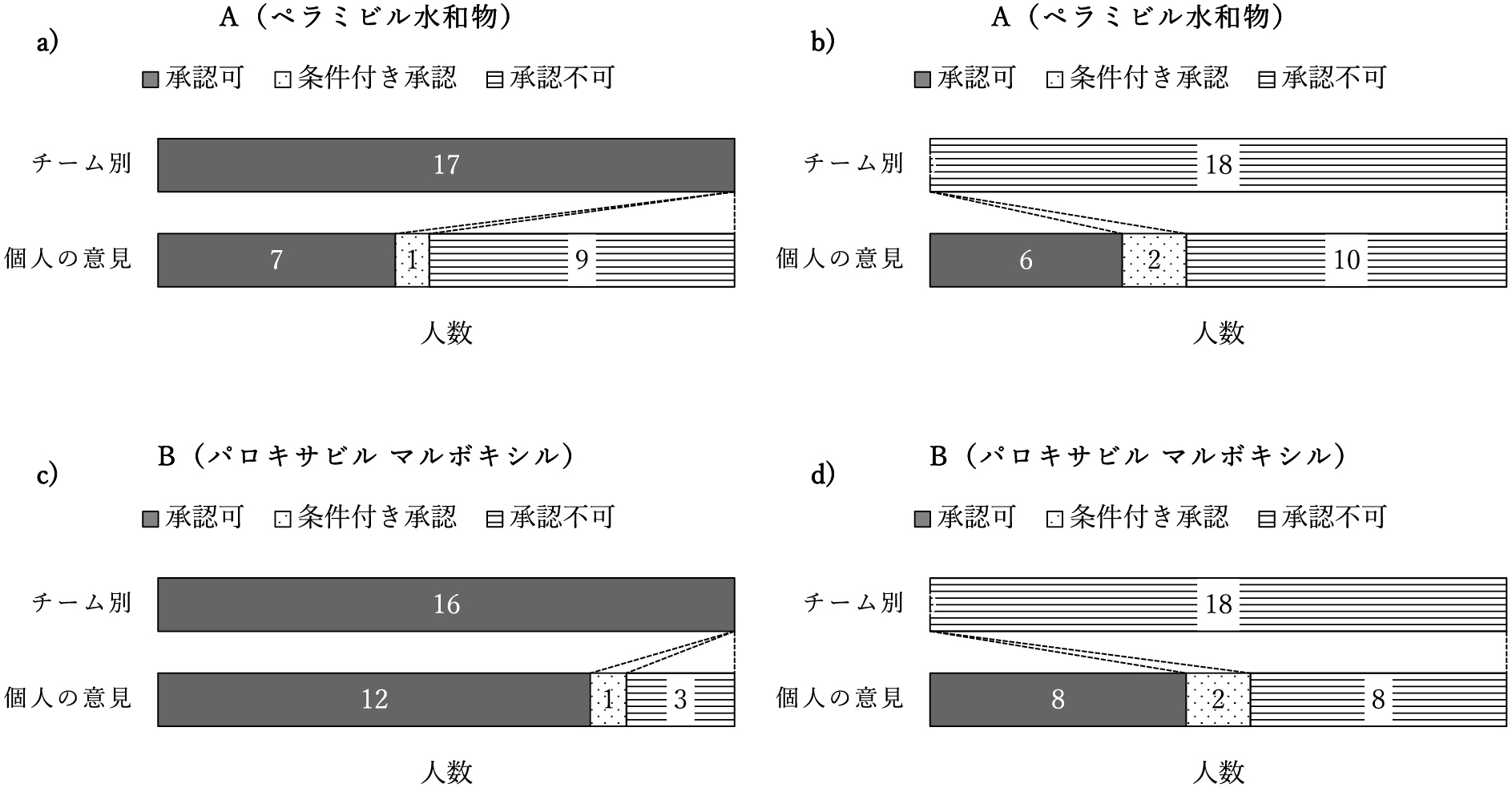
承認の可否に関する学生の意見
医薬品候補A(ペラミビル水和物)について,承認可グループ(図1a)17名の学生のうち,承認可は7名,承認不可は9名,効能を変更した上で承認可が1名であった.また,承認不可グループ(図1b)18名の学生のうち,承認可は6名,承認不可は10名,効能を変更した上で承認可が2名であった.
医薬品候補B(パロキサビル マルボキシル)について,承認可グループ(図1c)16名の学生のうち,承認可は12名,承認不可は3名,適用を狭め小児を除いた上で承認可が1名であった.また,承認不可グループ(図1d)18名の学生のうち,承認可は8名,承認不可は8名,条件付き承認が2名であった.具体的には,12歳未満は使用不可,情報を増やした後に承認可とする,小児は不可,成人も耐性について注意喚起すべき等の意見であった.
以上のように,ペラミビル水和物では,個人の意見として承認不可の学生が多かったが,パロキサビル マルボキシルでは,承認可の意見が一定程度を占めた.また,承認可グループから個人の意見を承認不可とした理由について,ペラミビル水和物の場合は,「判断できない.」,「有効性・安全性に関する追加資料が必要」,「B型の被験者が少ない.」,「第III相臨床試験でプラセボと比較していないことから有効性が不確実」,「重症患者に反復投与ではリスクが高くなるため不可」,「市販後に臨床試験の対象外の患者層に対する調査が必要」等であった.一方,パロキサビル マルボキシルの場合は,「国際共同第III相試験において,B型インフルエンザウイルス感染症に対する有効性が示されていない.」,「民族差が認められた.」,「小児における症例数が限られている.」,「本剤を服用した小児から耐性ウイルスが検出されたという報告がある.」,「追加の臨床試験を実施し,安全性が確保されてから承認すべきと考える.」,「症例数が少ない.」及び「12歳未満の患児を対象とした国内第III相試験では対照群を置いていない.」等であった.
3.模擬審査に関する学生の感想模擬審査に関する学生の感想の一部を抜粋し,表3に示した.承認審査に関する感想が最も多く(延べ57名),次いで模擬審査(同45),医薬品開発(同39)であった.データの解釈や判断を下すことの難しさ,審査プロセスについての理解,客観的評価の重要性等に関する感想もあった.
| 感想 | 延べ人数 |
|---|---|
| 医薬品開発について | 39 |
| ・多くの治験に基づき承認されていることを知り,大変さがよく分かった.薬について冷静に判断し,評価する能力を養っていきたい. | |
| ・承認されるためには臨床試験結果に精度が求められ,新薬として世に出ることの難しさの一端を知った. | |
| 承認審査について | 57 |
| ・薬を世に出すためには様々な観点から慎重に見極める必要があり,承認の可否は明確な証拠をもって判断すべきと思った. | |
| ・判断を下す難しさを感じた.どの程度なら承認可とするのか判断に迷った. | |
| ・データのどこを重く受け止めるか,どのように読み解くかによって承認の可否は変わってしまうと思った. | |
| ・手分けしたパートのみでなく,全体を把握し,詳細を考察したうえで,再び全体を俯瞰するという鉄則を徹底していきたい. | |
| 臨床試験について | 28 |
| ・臨床試験のデータを読み,そのデータが示す意味を理解し,その取扱いを考えるのが難しかった.評価方法(註:審査基準)を事前に決めておくべき. | |
| ・パロキサビル マルボキシルは新規作用機序の薬剤であり,オセルタミビルのみとの比較では難しいのではないかと感じた. | |
| ・耐性株出現について,小児の症例数が少なく判断が困難.もっとデータがあったほうが良い.小児,妊婦,民族差等のデータは集まりにくい. | |
| ・季節により流行する亜型が異なり,患者数が少ない場合があることから,A型,B型に効果があるかを評価するのは難しい. | |
| 安全性・市販後等について | 7 |
| ・臨床試験における安全性等の結果から添付文書の記載を考える必要があり,大変だと思った. | |
| ・既承認薬であっても,症例数の不足など承認不可としても良いと考えられる要素もみられた.必ずしも完璧な状態で承認され市場に出てくるわけではなく,市場に出てから製造販売後調査や市販直後調査等の情報収集を行い,医療現場への情報提供を行う必要があることがわかった. | |
| 模擬審査に関して | 45 |
| ・反対意見や指摘等を聴くことで,様々な角度からの見解を知ることができ面白かった.グループ内でも意見が異なり,新たな発見に繋がった. | |
| ・審査のプロセスや評価すべき項目について理解できた.また,客観的に評価を行う重要性も感じた.やりがいを感じた. | |
| ・身近な病気の治療薬が課題だったので,内容を理解しやすかった.承認不可となった品目のデータで課題をしてみたい. | |
| ・1グループの人数を少なくしても良いと思った(意見をまとめるのが難しかった). | |
| その他 | 25 |
| ・批判的な吟味を行う必要性を理解した.データを読み解くのに知識が必要.本質を見極める力をつけたい. | |
| ・パロキサビル マルボキシルは2018年に販売されたが既に耐性菌の報告もあるので,今後現場でどのように対応されていくのか興味がある. | |
| ・今まで暗記して覚えたことの意味を考える機会に恵まれ良かった.「座学,レポート提出」よりも充実していた. | |
| ・将来,薬剤師として責任を持って行動していかなければいけないと思った. |
2018年度は,履修生(4回生)62名を8グループに分け,審査対象品目ごとに申請者側と審査側の2グループで模擬審査を行った(表4).模擬審査の対象は既承認4品目(テリパラチド,ラニナミビルオクタン酸エステル水和物,フェブキソスタット,カナグリフロジン)であった.審査対象品目に関する情報(審査報告書,申請資料概要,添付文書等)は,学生がPMDAホームページの医療用医薬品情報検索サイト3) から収集・検討し,模擬審査に関する発表は申請者側と審査側で討論する形式とした.討論のために学生は審査報告書を熟読しなければならなかったため,一定の学習効果は認められた.しかしながら,発表はPMDAによる審査報告書に記載された内容の再現となり,学生自らが考えることや論理的思考力の向上への寄与等は十分ではなかった.そのため,2019年度は模擬審査対象の品目名を伏せ,承認の可否別にグループを分けて実施した.その結果,学生自らが考えた発表は,論点が絞られ全グループが時間内に終了した.発表に至る過程で活発な議論を行うこと等により,全員が模擬審査に携われたことも改善された点であった.学生自ら臨床試験のデータを様々な視点から検討しており,学生の感想には,「批判的な目でみることの必要性・重要性が理解できた.」,「承認の可・不可双方の意見や見方を知ることができ有益であった.」等があり(表3),論理的思考の向上もみられたと考えられたことから,模擬審査実施の意義があったと考える.また,対象品目の臨床試験成績等に基づく検討は,実際には学生自らがグループ内で項目ごとに分担して実施しており,活発な討論で多くの意見が出たためと考えられるが「1グループの人数が17~18人と多かったため,意見をまとめるのが難しかった.」との意見があった(表3).グループの構成人数を減らすのみではグループ数が多くなり,発表時間が不足する可能性があるため,模擬審査対象の品目数や検討項目を減らす等の工夫が必要と考える.
| 2018年度 | 2019年度 | |
|---|---|---|
| 履修生(4回生) | 62名 | 70名 |
| 1グループの人数/グループ数 | 7~8名/8グループ | 17~18名/4グループ |
| グループ分け | 申請者側・審査側 | 承認可・不可 |
| 課題数 | 4課題:テリパラチド,ラニナミビルオクタン酸エステル水和物,フェブキソスタット,カナグリフロジン | 2課題(既出):ペラミビル水和物,パロキサビル マルボキシル |
| 品目名・審査結果の事前認知 | 有り | 無し(医薬品候補物質として提示) |
| 資料の概要 | 検索サイトから学生が収集 | 教員が概略を提示 |
学生の模擬審査に関する感想として,「手分けされたパートのみでなく,全体を把握し,各パートの詳細を考えたうえで,再び全体を俯瞰するという鉄則を徹底していきたい.」,「審査については,資料全体をみなければいけないと思った.」,「安全性,有効性,用法・用量など,すべての項目が関連しているため,総合評価をする必要があると学んだ.」,「班内で項目別に分担作業を行い,円滑な作業の後に情報共有できたが,分担者作成のスライドでグループディスカッションを行ったため,各データの細かい比較がおろそかになってしまった.」,「多くのヒトの意見をまとめて承認の可否を決定するのは難しいであろうと感じた.」,「今回は手分けして行ったが,この内容をすべて網羅しようと思ったらとても時間のかかることだろうと思った.」等があった.このため,承認審査は全体を検討した上でなければ適切な評価を行うことはできないことについても,理解できたのではないかと考える.
また,2018年度の模擬審査の対象は,「国際共同治験が実施された品目」や「承認効能が申請時から変更された品目」等を選択したため様々な疾患領域の医薬品が対象となったが,2019年度は同一の疾患領域の医薬品を選択し,学生が身近に感じるインフルエンザ治療薬を対象品目とした.この場合は,当該疾患やその既存の治療薬について一定程度の知識及び情報を学生が有していたため,品目名を伏せた状態でも臨床試験成績に基づく承認の可否の検討が可能であったと考える.新有効成分含有医薬品の承認審査では,まず,開発された医薬品の候補物質がどのような疾患領域をターゲットとしているか,既承認薬がある場合にはそれと比べてどのような位置づけになることを期待しているのか,特徴(剤形,持続時間,投与間隔等)は何かなどについても検討する必要がある.したがって,ターゲットとする疾患の病態(大人と小児の違い等を含む)やその疾患に対する既存の治療方法及び薬物療法(国内外を含む.既承認薬の有無)等について知っていなければ,適切な審査はできない.今後,例えば糖尿病用薬など,学生にとって身近ではないかもしれないが患者数の多い疾患治療薬を題材として模擬審査を実施する際には,疾患とその治療法や薬物療法等の関連情報を説明してから実施する必要があると考える.
本学では本報告に示したように,模擬審査の実施は学生がRSについて理解する機会となったと考える.また,模擬審査の実施について,学生のレポートにおいて前向きな感想が得られたことから,今後も実施方法等を適宜改善した上で模擬審査の実施を継続したい.今後の実施方法等の改善事項として,発表の評価についてはルーブリック表を作成した上で実施すること,「承認の可否に関する個人の意見とその根拠・理由」を報告するタイミング等を検討する予定である.また,発表終了後に提出するレポートの課題の1つが「担当品目について個人の意見を述べること」であったため,口頭で審査報告書を読むことが必要であることを伝えていたが,全員が読んでいたとは言い難かった.このため,レポート課題の提示の方法についても改善したい.
本研究を行うにあたり,仔細にわたってご指導いただきました,一般社団法人日本QA研究会会長 平山佳伸先生(立命館大学薬学部 前教授)に深く感謝申し上げます.
発表内容に関連し,開示すべき利益相反はない.