Abstract
ヒトゲノム計画やその後の計測技術の進展にともない、多くの遺伝情報の取得・解析が容易になった。さらに、電子カルテやウェアラブルテクノロジー、大規模コホート研究の拡大によって、より多くの表現型のデータも得られるようになった。これら、遺伝型・表現型の両方におけるデータの拡大によって、より幅広い研究課題に取り組む絶好の機会が訪れている。人類遺伝統計学は、このようなデータを活用して、疾患などヒトの諸形質に影響を与える遺伝的変異をより深く理解するための強力なアプローチとなりうる。本稿では、ゲノムワイド相関解析やフェノムワイド相関解析、ポリジェニック・リスク・スコアなど、代表的な解析手法を取り上げ、複数の表現型の情報を考慮した遺伝情報解析など、近年の話題についても紹介する。
人類遺伝学における遺伝型・表現型データの拡大
遺伝学とは、表現型の多様性とその遺伝基盤を考える学問である。本年で、ヒトゲノム計画によるドラフトゲノム公開[1, 2]から20年となるが、その間に塩基配列決定技術は飛躍的な進歩をとげた。とくに、次世代シーケンサー技術の成熟[3]により、DNAシーケンシングのコストが100万分の1以下に低下した。また、HapMap project[4]や1000 Genomes Project[5]などにより、ヒトの集団内にどのような遺伝的多様性が存在するかが解明され、ありふれた遺伝的変異(common variants)、とくに一塩基多型(single nucleotide polymorphisms, SNPs)、を調べるマイクロアレイが用いられるようになった。これら、計測技術の進歩により、多くのDNAサンプルの遺伝的多様性を測定することが可能になった。
遺伝的多様性の計測技術の進歩とあわせて、表現型の計測技術も大きな変化をとげた。電子カルテが多くの病院に導入され、様々な疾患に関する診断結果、血球検査・尿検査などのバイオマーカーに関する情報、放射線科医・病理医により撮像された画像、薬剤の処方箋などの情報が集められている[6]。さらに、ウェアラブルデバイスにより記録された運動量のように、病院の外においても、健康に関する情報の収集・集積が行われている[7]。プライバシー保護や、データの所有権、異なるシステム間での情報交換を行うための基盤づくり(interoperability)など、解決されるべき重要な問題は多くあるものの、今後も表現型データは拡大を続けることと思われる。
さらに、いくつものリスク要因をもちうる複雑形質(complex traits)に影響を与える環境要因や遺伝的な要因を調べるため、世界中でいくつもの大規模コホート型研究が行われるようになった[8, 9, 10]。それぞれの研究デザインには違いがみられるものの、おおむね数十万人規模の大きさで、マイクロアレイやシーケンシングを用いた遺伝的変異の計測と、電子カルテ情報や質問票などによる表現型情報の収集などを組み込んだスタディであることが多い。日本では、バイオバンクジャパン[11]や東北メディカルメガバンク[12]の実施例がよく知られている。諸外国では、英国のUK Biobank[10, 13, 14]、フィンランドのFinnGen[15]、アメリカのMillion Veteran Program[16]やAll of Us Research Program[17]などが知られている。
このように、人類遺伝学の研究に必要なデータが、遺伝型・表現型の両方において多くのサンプルから集められるようになったことで、以前ではサンプルサイズの限界などからアプローチが困難であった問題を研究する機会が訪れているといえよう。
ゲノムワイド相関解析(GWAS)
表現型の多様性に影響を与える遺伝的変異を探索する手法には、家系情報を用いた連鎖解析や、ゲノムワイド相関解析(genome-wide association studies, GWAS)がある1。とくに、複雑形質(complex traits)の解析には、統計的検出力(statistical power)の観点から、ゲノム上の連鎖不平衡ブロックの多くをカバーするように設計された遺伝的マーカーに対して遺伝型を調べ、ゲノムワイド相関解析を行うことが提唱され、2000年代から実施されている[18, 19, 20, 21]。ゲノムワイド相関解析では、解析に用いられる遺伝的変異それぞれについて、特定の形質に対する線形回帰モデルを考え、回帰分析を行う2。
|
式(1)
y
~
age
+
sex
+
genotype PCs
+
G
{y "~" "age" +"sex" +"genotype PCs" +G}
|
-
ここで、yは解析の対象としたい量的形質、Gはひとつの遺伝的変異となる。回帰分析に年齢(age)や性別(sex)などの共変量(covariates)を組みこむことで、これらに交絡するような相関を検出することを避けることが一般的である。スタディデザインによっては、年齢の二乗項(age2)や、ボディマス指数(body mass index, BMI)などを共変量に組み込むこともある。Genotype PCsというのは、遺伝的変異データの行列(個人×遺伝的変異のサイズをもつ)に主成分分析(principal component analysis, PCA)を適用したとき各個人について得られる主成分得点のことで[22]、集団構造(population structure)をコントロールするための項としてよく用いられる[23]。もちろん、これらの共変量を組み込むだけで、品質の高い相関解析を実施できるというわけではなく、遺伝型データや表現型データのクオリティ・コントロールも重要である[24]。回帰分析を行うことで、遺伝的変異Gに対する効果量(β)の推定値(
β
^
{ widehat β}
)やその標準誤差(
SE
(
β
^
)
{SE \( { widehat β} \)}
)、p値などの要約統計量(summary statistics)が得られる3(図1)。GWASでは多くの遺伝的変異を解析するため、多重検定補正を行い、p<5×10−8をゲノムワイド有意水準とするのが一般的である。GWASに用いられるサンプルサイズが限られていたころは、このゲノムワイド有意水準に達する、推定効果量の小さい相関を発見することは困難であったが、先に述べた計測技術の進展により多くのサンプルを対象とした研究が推進されることになり、身長・肥満・糖尿病など多くの複雑形質において、ゲノムワイド有意水準に達する多くの相関が同定された[25, 26, 27]。遺伝的な影響を与える遺伝的変異・座位の種類や数は、表現型によって異なり、多様な表現型それぞれについて遺伝的基盤を解明する研究が続けられている。ひとつの形質に複数の遺伝子座が影響を与えることを、ポリジェニシティ(polygenicity)とよぶ[28, 29]。
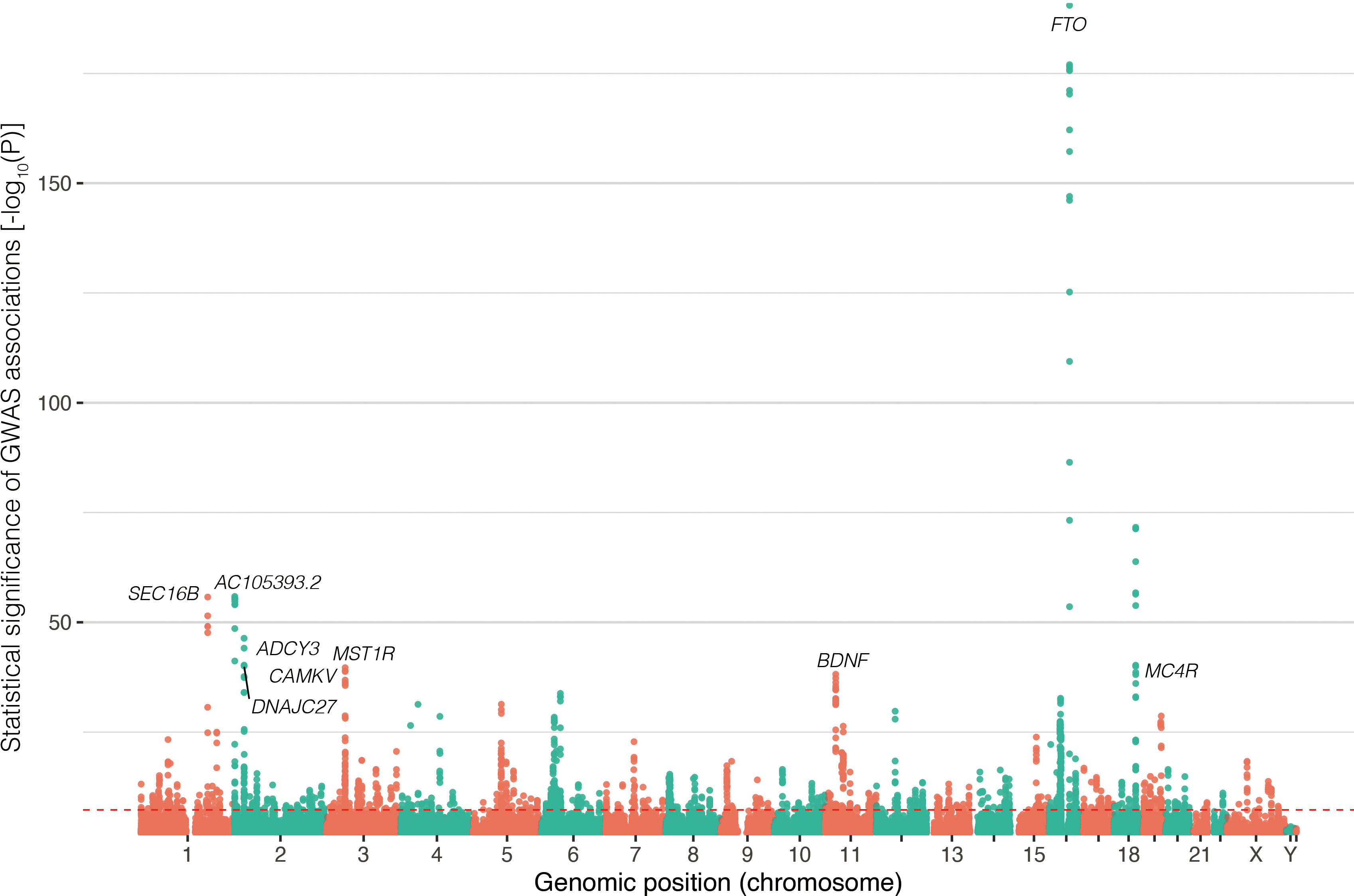
式(1)では、量的形質を対象とした線形回帰式を示したが、疾患のように2値(疾患[ケース]またはコントロール)をとる表現型については、線形回帰のかわりにロジスティック回帰を適用すればよい。これらに加え、一般化線形モデルの枠組みで、生存時間解析のCox's proportional hazard model[30]を用いて、疾患の発症時期に対してゲノムワイド相関解析を用いる試みも行われている[31, 32, 33]。また、近親者(related individuals)を含めて相関解析を行う場合は、線形混合モデル(linear mixed model)を用いることが一般的である[34]。さらに、複数のスタディで解析されたGWASの結果を組み合わせる、メタ・アナリシスの手法も開発され多くの研究に応用されている[25, 26, 27, 35, 36]。GWASの結果は、インタラクティブなデータブラウザとして提供されているほか[37, 38]、GWAS catalogというデータベースに集められており[39, 40]、Open Targets Genetics[41]などのプラットフォームから利用できるようになっている。なお、近年、GWASなど遺伝情報解析に用いられるサンプルの多様性の欠如について問題提起がなされており、ヨーロッパ系以外のサンプルを解析に用いることへの関心が高まっている[42, 43, 44]。
これらは、一般化線形モデルを用いた回帰分析という比較的シンプルな統計解析手法の応用であるが、サンプルサイズの増加などに伴う計算量の増加から、スパース性をうまく利用した新しいデータフォーマットの提案・より効率的な計算法の実装・近似法の導入など、近年も新規の手法が提案され続けている[45, 46]。GWASのように10年以上用いられているような基盤的な解析法の改良は、多くの研究にインパクトをもたらす魅力があることを記しておきたい。
表現型データの取得(phenotyping)
遺伝的情報の取得とクオリティ・コントロールの手法が標準化されるなかで、表現型データの取得(phenotyping)の重要性が相対的に高まっていると考えられる。たとえば、電子カルテ・医療情報から疾患の診断情報を網羅的に取得するような手法が開発され、データ欠損やバイアスを検出・補正するようなデータ・モデリング研究が行われている[47]。GWASでは、一つの疾患を選び、ゲノム全体のSNPsなどの遺伝的変異に対して相関解析を行うが、一つの遺伝的変異に注目し、観測された表現型全体(phenome)に対して網羅的な相関解析を行う、Phenome-wide association studies(PheWAS)というアプローチも用いられている(図2)[48]。バイオバンクなど、多くの表現型と遺伝情報が両方計測されているようなコホートにおいて、GWASとPheWASの両方の解析が行われることも珍しくなくなってきた。PheWASが進行するにつれて、一つの遺伝子座が複数の形質に影響を与える、多相遺伝(pleiotropy)という様子が多く観察されるようになった[49]。
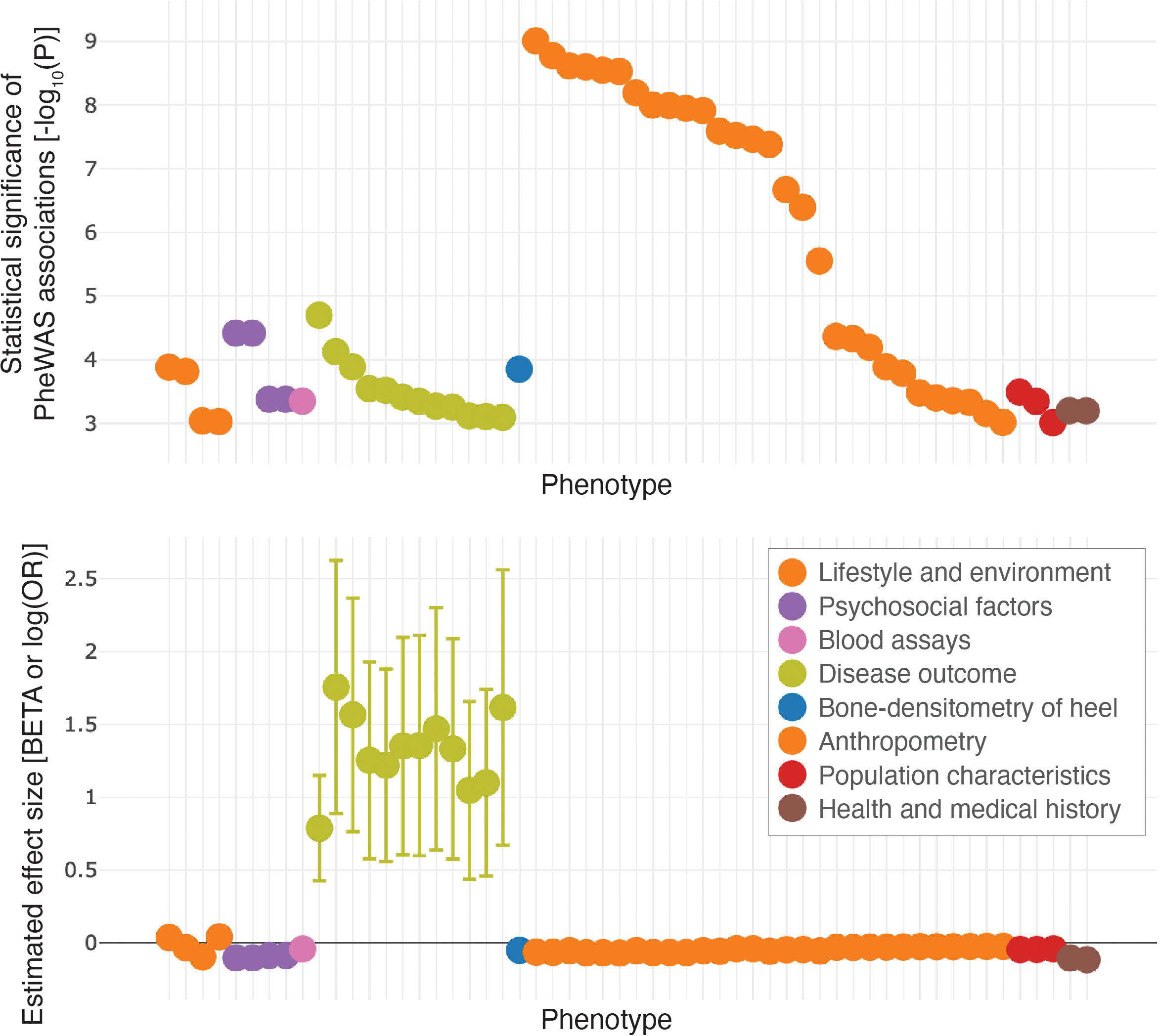
表現型データは様々なデータソースから取得できる。発症年齢が遅い疾患の遺伝基盤を探索するには、研究参加者のスタディ参加時点での疾患の状態を用いてGWASを行うかわりに、疾患の家族歴を将来発症するかもしれない疾患状態の代理変数として用いたほうが、より多くのケースを得られて検出力が高まるかもしれない[50, 51]。また、一人の表現型の情報は、電子カルテに加え、質問票などにも含まれているかもしれない[13, 14]。複数のデータソースには、ランダムではない情報欠損などが含まれているため、異なるデータソース間の一致度を表現型のレイヤーで直接比較することは困難である。我々も、英国のUK Biobankのデータを用いてこのようなデジタル・フェノタイピング(digital phenotyping)の問題を考え、病院内の疾患診断コードや質問表、家族歴など、それぞれの情報源を用いてGWASを行い、その結果を比較することで一致度を評価できること、また複数の情報源を組み合わせることで検出力を向上できることなどを報告した[52]。複数の情報源からの表現型の取得、クオリティ・コントロール、その組み合わせの方法論などについては、データ取得がどのように行われているかなどの状況を加味して、スタディごとに検討を行う価値があると考えられる。
今後、より多くのクリニカルサンプルから遺伝的情報が取得されるにつれて、バイオインフォマティクスとクリニカルインフォマティクスの両方の分野の協力による研究が進展すると考えられる。
希少な遺伝的変異に対する相関解析
GWASやPheWASにおいて検定にかけられる(遺伝型、表現型)ペアの数が増えることで、多重検定の補正を適切に行わなければいけなくなる。一般に、遺伝的変異の数(~106)に対して表現型の数は少ない(~103)こと、バイオバンクなどの大規模コホートではサンプルサイズが十分に大きいことを考えると、ありふれた疾患(common disease)に対するありふれた遺伝的変異(common variant)の相関を検定している場合には、統計的検出力(statistical power)は、さほど大きな問題とならないかもしれない。
しかし、希少変異(rare variant)の解析は困難で、統計的検出力の問題が生じる[53, 54]。同一遺伝子上の希少変異に観測される相関の効果をまとめて、burden test[55, 56](効果の和に注目する検定)やvariance-component test[57, 58](効果の分散に注目する検定)を行う方法、これらの組み合わせる方法[59, 60]など、様々なアプローチが提案されている[53, 54]。ボトルネック効果(population bottleneck)など、ユニークな歴史をたどった創始者集団(founder population)では希少変異がエンリッチしていて、他の集団では検出が難しいような遺伝的相関を検出できることもある[61]。また、複数の遺伝的変異、複数の表現型、複数のスタディの情報をベイズ統計の枠組みで組み合わせるような方法も、近年提案されている[62]。今後、大規模集団において得られたエクソームシーケンシングや全ゲノムシーケンシングの解析が進むなかで、様々なインフォマティクス手法の開発がさらに進展することが期待される。
GWASの結果を活用した二次解析
GWASによって得られる要約統計量(summary statistics)は、それ自体が疾患や非疾患形質を研究するための有力な手段となりうるが、さらなる下流解析の入力として用いられることも多い(図3)[63]。先に紹介した、メタ・アナリシスによって、複数のコホートでのGWAS解析からロバストな相関を探索する手法や、Fine-mappingという解析手法によって連鎖している遺伝的変異の中から原因遺伝的変異を同定する試み[64]は、近年よく用いられている。また、GWASで得られたゲノムワイド相関と、遺伝子アノテーションモデルや大規模エピジェネティクデータ、あるいは発現データやオントロジーなどに対してエンリッチメント解析を行う試みもなされてきた[65, 66, 67, 68]。このようなエンリッチメント解析の難点は、非コード領域上の相関をどのように既存の機能データに結びつけるかという方法論と、遺伝的変異間の連鎖の構造の取り扱いであった。少なくとも後者の難点は、LD score regression(LDSC)という手法の開発など[69, 70, 71]、解決の方法が示されつつあると考えることができるであろう。

さらに、LDSCなどの手法を用いて、GWASの要約統計量から遺伝率[72]や遺伝的相関[73]といった遺伝学におけるパラメータ推定を行うことも可能となった[69, 70, 71](Box 1)。遺伝率(heritability)とは、遺伝型(の加法的な効果)で説明できる表現型の分散が表現型全体の分散に占める割合として定義される量で、集団内の表現型のばらつきに対する遺伝的な効果を定量したものである[72]。「広義の遺伝率(H2)」と加法的な効果に限定した「狭義の遺伝率(h2)」という異なる概念が存在するほか、数多くの誤謬が知られているため、詳しくは専門誌のレビューなどを参照していただきたい[72]。一方、遺伝的相関(genetic correlation, rg)とは、2つの形質に影響を与える遺伝的な効果が、どの程度共通しているかを定量したものである[73]。2つの形質の間で共有される多相遺伝(pleiotropy)の程度をゲノム全体で調べた結果を表す量と考えるとわかりやすいかもしれない。遺伝率や、遺伝的相関は、実際に観測することは困難で、統計モデルによって推定される値であることに注意してほしい。

遺伝的相関を調べることで、複数の複雑形質が共通の遺伝的基盤を持っていることが明らかになるなど、疾患やその他の形質に関する理解が深まることがある[73]。遺伝的相関は、ゲノム全体における共通の遺伝相関の程度を1つの数値としてまとめたものであるので、実際にどのような遺伝的領域が共通した効果を持つのかは明らかではない。この疑問に答えるため、local genetic correlationといった、特定のゲノム領域のみについて遺伝的相関(genetic correlation)を評価するような手法が開発された[74]。我々も、同様の問題に対する異なるアプローチとして、Decomposition of Genetic Associations(DeGAs)という手法を提案した[75]。DeGAsでは、複数の形質に共通する、直接は観測されていない遺伝基盤のモジュールが存在すると考え、これをGWASとPheWASによって得られる要約統計量の行列の特異ベクトル・隠れ要素(latent components)として抽出する。論文では、隠れ要素を複雑形質のGWAS結果の解釈や実験的検証のための候補遺伝子探索に活用する方法を示した。このような遺伝的基盤の分解(decomposition)の手法は、次項で述べるようなポリジェニック・リスク・スコアの解釈可能性を向上させるために用いられたり[76]、ベイジアン縮小推定を用いた方法に発展させられたり[77]、複数コホートのメタ・アナリシス結果の解析に応用されたりしている[78]。今後は、メンデリアン・ランダマイゼーション(Mendelian randomization)といった、因果推論の枠組みとあわせて考えることで、複雑形質の原因となる遺伝的基盤の同定などの進展が期待される。
ポリジェニック・リスク・スコア(PRS)
先に述べたGWASは、疾患やその他の表現型に相関を示す遺伝的変異の探索に用いられてきた。GWASに用いられるサンプルサイズが増加するにしたがって、これらのGWASの結果から、個人の遺伝的な疾患リスクを予測するためのリスクスコアを計算することへの関心が高まった。多くの遺伝子座の効果により、複雑な遺伝形質を予測する手法として、ポリジェニック・リスク・スコア(polygenic risk score, PRS)と呼ばれる方法が提案されている[28, 79, 80, 81, 82, 83]。PRSモデルは、線形モデルとして表されることが多い。
|
式(2)
PRS
i
=
∑
j
β
^
j
X
i
,
j
{"PRS"_i ={ sum csub j {{ widehat β}_j X_{i , j}}}}
|
-
ここで、PRSiとは、PRSモデルにより予測される個人iに関するスコア、βjは多変量回帰分析モデルにおける効果量(真の値はわからないので、推定値
β
^
j
{ widehat β}_j
が用いられる)、Xi,jは個人iの遺伝的変異jにおけるリスクアレルの数(0、1、2、あるいは欠損値[NA]のどれか)を表す行列である。欠損値の取り扱いは様々で、遺伝型インピュテーション(genotype imputation)により補完されることもある[83]。GWASにおいては、それぞれの遺伝的変異を独立に取り扱い回帰と検定を行うため、GWASで有意水準を超えた遺伝的変異とその単変量解析での推定効果量を、遺伝的変異同士の連鎖構造を考慮せずにそのまま使うことは難しいことには注意が必要となる。
このようなPRSモデリングは、古くから農業分野での育種などにも使われてきた[84]。人類遺伝学においては、サンプルサイズの増加や変数選択・効果量推定の手法開発の進展により、モデルの予測性能が向上して近年注目を集めている[81, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94]。ポリジェニック・リスク・スコアを用いて複数の遺伝的変異の効果を組み合わせることで、集団の中から、遺伝的要因により疾患リスクが平均よりも高くなっている人を同定することができる。モデルの予測性能の向上により、単一遺伝子疾患に関する保因者のスクリーニングを行うように、ポリジェニック・リスク・スコアを用いて複雑形質をスクリーニングすることへの期待が寄せられていたり[88]、従来から単一遺伝子疾患として知られていた疾患にも、ポリジェニックなリスク成分が存在することが報告されていたりしている[95]。GWASと同様に、PRSモデル構築に用いられるサンプルには多様性が欠如していることが指摘されており、トレーニングに使われた集団以外へ用いられた場合に精度が落ちる点には注意が必要である[42, 43, 96, 97]。
PRSモデリングは、高次元なスパースデータを用いた回帰分析の問題とも考えられるため、統計・機械学習の方法の応用や手法開発も行われている。たとえば、ベイジアン多変量解析の枠組みで、GWASやそのメタ・アナリシスから得られる要約統計量と遺伝的変異の連鎖不平衡(linkage disequilibrium, LD)の強さを表したLD matrixを用いて、ベイジアン線形モデルを学習する手法などが提案されている[90]。レファレンスに用いられるLD matrixを縮小(shrink)させることで、スパースなLD matrixを構築し利用する手法も提案されている[87, 90, 98]。また、LDではなく特徴量のsparsityを仮定するpenalized regression modelの枠組みで、Lasso (L1 penalized) regression、Ridge (L2 penalized) regression、Elastic netを用いる方法も提案されている[91]。このような正則化項つきの回帰モデルをフィットするには、通常、個人レベルのデータをメモリに読み込むことが必要となる。しかし、バイオバンクスケールの大規模データ(n>300,000、p>500,000)を読み込むのは、困難なこともある。幸いにもLasso regressionの場合は、変数選択(feature selection)を効率的に行うためのスクリーニング・ルールが研究されており、我々も統計の専門家との共同研究を通じて“strong rule”[99]を応用したBatch Screening Iterative Lasso(BASIL)という手法を提案し、R snpnetパッケージとして実装した[91]。また、遺伝的変異データのスパース性を活用してメモリ使用量を削減する取り組みも行っている[94]。
先の遺伝的相関のところでみたように、複数の形質が共通の遺伝基盤を持つ場合がある。このような場合は、ポリジェニック・リスク・スコアのような予測モデルの枠組みでも、複数の形質の情報を考慮した解析を行うことが有効であろう。我々は、血液や尿中のバイオマーカーの包括的な遺伝的解析のなかで、これらのバイオマーカーに対して構築されたポリジェニック・リスク・スコアの予測モデルを、疾患に対して構築されたポリジェニック・リスク・スコアの予測モデルと線形に組み合わせることで、疾患の予測精度が向上する例をいくつか示した[100]。これは、精度のよいポリジェニック予測モデルの開発には、サンプルサイズの増加と、複数形質を考慮した解析が、相互補完的に寄与できることを示唆している。また、先に述べた、Batch Screening Iterative Lasso(BASIL)というような正則化つき多変量回帰を複数形質の状況に拡張した、Sparse reduced rank regression(SRRR)という手法を、統計の専門家との共同研究により開発した[101]。SRRRのモデルは、L1正則化項によるスパース性に加えて、複数の遺伝的変異から複数の表現型への回帰係数をあらわす行列のランクに制約をつけるため、DeGAs[75]などに見られるようなdecompositionの手法のmultivariate-multi-responseへの拡張にもなっている。Batch Screening Iterative Lasso(BASIL)[91]やSparse reduced rank regression(SRRR)[101]の手法は、生存時間解析のモデルにも拡張され、疾患の発症時期に関わるポリジェニック・リスク・モデリングにも活用されはじめている[92, 102]。
ここまでの、ポリジェニック・リスク・スコアの手法の紹介では、すべて線形モデルを用いたものを取り上げた。複数の遺伝子座同士の交絡作用や、各座位における加法的ではない効果などは、どのように扱えばよいのだろうか。2型糖尿病の遺伝基盤では、遺伝子座同士の交絡作用は見られなかったというような研究はあるものの[103]、これが他の多くの複雑形質にも見られる特徴なのかは未だによくわかっていない。データと計算資源の増加に注目し、線形よりも表現力が高いモデルを用いて、ポリジェニック・リスク・スコアを構築するような試みは行われるであろう。このようなモデルの評価においては、トレーニングに使われなかったコホート(とくに異なる祖先グループに属する人々からなるコホート)においても、予測精度の向上が見られるかを調べることが重要になるのではないかと予想する。
まとめ
本稿では、人類遺伝統計学の大規模情報解析について、代表的な手法と近年の進展についてとりあげた。シーケンシング技術の進展による遺伝的な情報爆発に加え、表現型のデータの蓄積が進んでいること、これにより人類遺伝学の様々な問題に取り組むことが可能になっていることを紹介した。遺伝型・表現型の両方のデータをもとに、ゲノムワイド相関解析(GWAS)やフェノムワイド相関解析(PheWAS)といった、単変量回帰分析が行われていること、それらの結果が二次解析に用いられていることを述べた。また、解析対象のサンプルサイズの増加にともなって、複数の表現型や遺伝的変異の間の相関構造を活用した、多変量解析の応用例が、ポリジェニック・リスク・スコア(polygenic risk score)などで盛んに活用されていることを示した。
今後は、バイオインフォマティクスや人類遺伝学に加え、クリニカルインフォマティクスや高次元統計学などの関連分野の交流により、研究分野のさらなる発展が期待される。筆者の個人的な見解では、次にあげるような課題の研究が(引き続き)重要となるであろう(図3)。
-
・網羅的な表現型情報の取得・表現のためのデータ基盤の構築。とくに、時系列クリニカルデータの継続的な取得と活用、あるいは表現型データの意味論(semantics)をオントロジーなどにより標準化する試みは重要な役割を果たすだろう。また、多様な表現型データのデータソースやスタディデザインごとの個別の事情を鑑みた、丁寧なクオリティ・コントロールも引き続き重要となると考えられる。
-
・大規模データを活用した、ゲノムワイド・フェノムワイド相関解析の実施と、データ共有。とくに、ヨーロッパ系以外のサンプルを用いた解析や、祖先グループが異なる集団でもロバストに再現されるゲノムワイド相関の同定、さらにはエクソーム・全ゲノムシーケンシングのデータなどのスパース性を活用するような計算手法の開発は、大きなインパクトをもたらすだろう。
-
・GWAS結果や、ポリジェニック・リスク・モデルの回帰係数など、要約統計量を入力としてとる二次解析の手法開発と応用。要約統計量には個人レベルの情報が含まれないため、個人情報漏洩のリスクは比較的少なく、また、サンプルサイズが増加しても要約統計量のデータサイズは増加しない。遺伝的情報と表現型情報のデータサイズが拡大を続けるなか、要約統計量を入力とした計算手法の開発は、スケーラブルな解析手法を開発するための現実的な解決策のひとつといえるだろう。
バイオインフォマティクスや関連分野の研究者の参入による、さらなる研究の進展や分野の発展を願って、本稿の結びとしたい。
謝辞
王青波氏、太田力文氏からは、ご多忙のなか原稿に目を通していただき、内容や表現に関する貴重なアドバイスを頂いた。ここに深謝の意を表する。もちろん、内容に誤りや不適切な点があれば、すべて筆者の責にあるのは言うまでもない。また、筆者は公益財団法人船井情報科学振興財団による海外留学奨学金と、Stanford University School of Medicineによりサポートを受けた。ここに深謝の意を表する。なお、本稿に書かれている内容は、筆者の見解によるもので、これらの資金提供者は、掲載されている内容や、投稿・出版の可否に影響を与えていない。
脚注
1 GWASは「ゲノムワイド関連解析」として訳されることが一般的である。GWASで調べられているのは、表現型と遺伝的変異の統計的な相関にほかならず、因果関係などその他の関係との混同を避けるため、本稿では「ゲノムワイド相関解析」の訳語を用いることとした。
2 式(1)には、R言語などに見られる記法を用いた。これは、次の式のように書き換えることもできる。
|
y
i
=
y
0
+
β
age
×
age
i
+
β
sex
×
sex
i
+
∑
k
β
PC
(
k
)
×
PC
(
k
)
i
+
β
G
×
G
i
+
ε
i
|
-
ここで、iは個人iに関する量であることを示す添字、y0は定数項、βは式(1)の右辺にある説明変数(独立変数)それぞれに関する効果量、そしてεiは誤差項である。
3 回帰分析を行うと、式(1)の右辺に含まれるそれぞれの説明変数(独立変数)について効果量の推定値・標準誤差・p値などの要約統計量が得られる。GWASを実施する際には、遺伝的変異Gに対する要約統計量に関心があるため、
β
G
^
を
β
^
と表記するなど簡略化した記法を用いた。
References
- [1] Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409: 860-921. doi:10.1038/35057062
- [2] Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291: 1304-1351. doi:10.1126/science.1058040
- [3] Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550: 345. doi:10.1038/nature24286
- [4] International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449: 851-861. doi:10.1038/nature06258
- [5] The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526: 68. doi:10.1038/nature15393
- [6] Jensen PB, Jensen LJ, Brunak S. Mining electronic health records: towards better research applications and clinical care. Nat Rev Genet. 2012;13: 395-405. doi:10.1038/nrg3208
- [7] Christle JW, Hershman SG, Torres Soto J, Ashley EA. Mobile Health Monitoring of Cardiac Status. Annu Rev Biomed Data Sci. 2020;3: 243-263. doi:10.1146/annurev-biodatasci-030220-105124
- [8] Gulcher J, Stefansson K. An Icelandic saga on a centralized healthcare database and democratic decision making. Nat Biotechnol. 1999;17: 620. doi:10.1038/10796
- [9] Hakonarson H, Gulcher JR, Stefansson K. deCODE genetics, Inc. Pharmacogenomics. 2003;4: 209-215. doi:10.1517/phgs.4.2.209.22627
- [10] Ollier W, Sprosen T, Peakman T. UK Biobank: from concept to reality. Pharmacogenomics. 2005;6: 639-646. doi:10.2217/14622416.6.6.639
- [11] Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, Kiyohara Y, et al. Overview of the BioBank Japan Project: Study design and profile. J Epidemiol. 2017;27: S2-S8. doi:10.1016/j.je.2016.12.005
- [12] Kuriyama S, Yaegashi N, Nagami F, Arai T, Kawaguchi Y, Osumi N, et al. The Tohoku Medical Megabank Project: Design and Mission. J Epidemiol. 2016;26: 493-511. doi:10.2188/jea.JE20150268
- [13] Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12: e1001779. doi:10.1371/journal.pmed.1001779
- [14] Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562: 203-209. doi:10.1038/s41586-018-0579-z
- [15] Mars N, Koskela JT, Ripatti P, Kiiskinen TTJ, Havulinna AS, Lindbohm JV, et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med. 2020;26: 549-557. doi:10.1038/s41591-020-0800-0
- [16] Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70: 214-223. doi:10.1016/j.jclinepi.2015.09.016
- [17] All of Us Research Program Investigators, Denny JC, Rutter JL, Goldstein DB, Philippakis A, Smoller JW, et al. The “All of Us” Research Program. N Engl J Med. 2019;381: 668-676. doi:10.1056/NEJMsr1809937
- [18] Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32: 650-654. doi:10.1038/ng1047
- [19] Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6: 95-108. doi:10.1038/nrg1521
- [20] The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447: 661-678. doi:10.1038/nature05911
- [21] Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017;101: 5-22. doi:10.1016/j.ajhg.2017.06.005
- [22] Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, et al. Genes mirror geography within Europe. Nature. 2008;456: 98-101. doi:10.1038/nature07331
- [23] Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38: 904-909. doi:10.1038/ng1847
- [24] Marees AT, de Kluiver H, Stringer S, Vorspan F, Curis E, Marie-Claire C, et al. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int J Methods Psychiatr Res. 2018;27: e1608. doi:10.1002/mpr.1608
- [25] Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45: 501-512. doi:10.1038/ng.2606
- [26] Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518: 197-206. doi:10.1038/nature14177
- [27] Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50: 1505-1513. doi:10.1038/s41588-018-0241-6
- [28] International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460: 748-752. doi:10.1038/nature08185
- [29] Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169: 1177-1186. doi:10.1016/j.cell.2017.05.038
- [30] Cox DR. Regression models and life-tables. J R Stat Soc. 1972;34: 187-202. doi:10.1111/j.2517-6161.1972.tb00899.x
- [31] Kapoor M, Wang J-C, Wetherill L, Le N, Bertelsen S, Hinrichs AL, et al. Genome-wide survival analysis of age at onset of alcohol dependence in extended high-risk COGA families. Drug Alcohol Depend. 2014;142: 56-62. doi:10.1016/j.drugalcdep.2014.05.023
- [32] Rizvi AA, Karaesmen E, Morgan M, Preus L, Wang J, Sovic M, et al. gwasurvivr: an R package for genome-wide survival analysis. Bioinformatics. 2019;35: 1968-1970. doi:10.1093/bioinformatics/bty920
- [33] Staley JR, Jones E, Kaptoge S, Butterworth AS, Sweeting MJ, Wood AM, et al. A comparison of Cox and logistic regression for use in genome-wide association studies of cohort and case-cohort design. Eur J Hum Genet. 2017;25: 854-862. doi:10.1038/ejhg.2017.78
- [34] Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47: 284-290. doi:10.1038/ng.3190
- [35] Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26: 2190-2191. doi:10.1093/bioinformatics/btq340
- [36] Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, Moon S, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582: 240-245. doi:10.1038/s41586-020-2263-3
- [37] McInnes G, Tanigawa Y, DeBoever C, Lavertu A, Olivieri JE, Aguirre M, et al. Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. Bioinformatics. 2018. doi:10.1093/bioinformatics/bty999
- [38] Gagliano Taliun SA, VandeHaar P, Boughton AP, Welch RP, Taliun D, Schmidt EM, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020;52: 550-552. doi:10.1038/s41588-020-0622-5
- [39] Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42: D1001-6. doi:10.1093/nar/gkt1229
- [40] MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45: D896-D901. doi:10.1093/nar/gkw1133
- [41] Ochoa D, Hercules A, Carmona M, Suveges D, Gonzalez-Uriarte A, Malangone C, et al. Open Targets Platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 2021;49: D1302-D1310. doi:10.1093/nar/gkaa1027
- [42] Kim MS, Patel KP, Teng AK, Berens AJ, Lachance J. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018;19: 179. doi:10.1186/s13059-018-1561-7
- [43] Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51: 584-591. doi:10.1038/s41588-019-0379-x
- [44] Green ED, Gunter C, Biesecker LG, Di Francesco V, Easter CL, Feingold EA, et al. Strategic vision for improving human health at The Forefront of Genomics. Nature. 2020;586: 683-692. doi:10.1038/s41586-020-2817-4
- [45] Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4: 7. doi:10.1186/s13742-015-0047-8
- [46] Mbatchou J, Barnard L, Backman J, Marcketta A, Kosmicki JA, Ziyatdinov A, et al. Computationally efficient whole genome regression for quantitative and binary traits. bioRxiv. 2020. doi:10.1101/2020.06.19.162354
- [47] Li Y, Nair P, Lu XH, Wen Z, Wang Y, Dehaghi AAK, et al. Inferring multimodal latent topics from electronic health records. Nat Commun. 2020;11: 2536. doi:10.1038/s41467-020-16378-3
- [48] Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26: 1205-1210. doi:10.1093/bioinformatics/btq126
- [49] Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, Polderman TJC, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51: 1339-1348. doi:10.1038/s41588-019-0481-0
- [50] Purcell S, Sham P, Daly MJ. Parental phenotypes in family-based association analysis. Am J Hum Genet. 2005;76: 249-259. doi:10.1086/427886
- [51] Liu JZ, Erlich Y, Pickrell JK. Case-control association mapping by proxy using family history of disease. Nat Genet. 2017;49: 325-331. doi:10.1038/ng.3766
- [52] DeBoever C, Tanigawa Y, Aguirre M, McInnes G, Lavertu A, Rivas MA. Assessing Digital Phenotyping to Enhance Genetic Studies of Human Diseases. Am J Hum Genet. 2020;106: 611-622. doi:10.1016/j.ajhg.2020.03.007
- [53] Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95: 5-23. doi:10.1016/j.ajhg.2014.06.009
- [54] Auer PL, Lettre G. Rare variant association studies: considerations, challenges and opportunities. Genome Med. 2015;7: 16. doi:10.1186/s13073-015-0138-2
- [55] Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat Res. 2007;615: 28-56. doi:10.1016/j.mrfmmm.2006.09.003
- [56] Price AL, Kryukov GV, de Bakker PIW, Purcell SM, Staples J, Wei L-J, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86: 832-838. doi:10.1016/j.ajhg.2010.04.005
- [57] Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89: 82-93. doi:10.1016/j.ajhg.2011.05.029
- [58] Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7: e1001322. doi:10.1371/journal.pgen.1001322
- [59] Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91: 224-237. doi:10.1016/j.ajhg.2012.06.007
- [60] Derkach A, Lawless JF, Sun L. Robust and powerful tests for rare variants using Fisher's method to combine evidence of association from two or more complementary tests. Genet Epidemiol. 2013;37: 110-121. doi:10.1002/gepi.21689
- [61] Tanigawa Y, Wainberg M, Karjalainen J, Kiiskinen T, Venkataraman G, Lemmelä S, et al. Rare protein-altering variants in ANGPTL7 lower intraocular pressure and protect against glaucoma. PLoS Genet. 2020;16: e1008682. doi:10.1371/journal.pgen.1008682
- [62] DeBoever C, Aguirre M, Tanigawa Y, Spencer CCA, Poterba T, Bustamante CD, et al. Bayesian model comparison for rare variant association studies of multiple phenotypes. bioRxiv. 2018. doi:10.1101/257162
- [63] Pasaniuc B, Price AL. Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet. 2017;18: 117-127. doi:10.1038/nrg.2016.142
- [64] Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19: 491-504. doi:10.1038/s41576-018-0016-z
- [65] Segrè AV, DIAGRAM Consortium, MAGIC investigators, Groop L, Mootha VK, Daly MJ, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6. doi:10.1371/journal.pgen.1001058
- [66] McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28: 495-501. doi:10.1038/nbt.1630
- [67] Pers TH, Karjalainen JM, Chan Y, Westra H-J, Wood AR, Yang J, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6: 5890. doi:10.1038/ncomms6890
- [68] Iotchkova V, Ritchie GRS, Geihs M, Morganella S, Min JL, Walter K, et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat Genet. 2019;51: 343-353. doi:10.1038/s41588-018-0322-6
- [69] Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47: 291-295. doi:10.1038/ng.3211
- [70] Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47: 1228-1235. doi:10.1038/ng.3404
- [71] Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47: 1236-1241. doi:10.1038/ng.3406
- [72] Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9: 255-266. doi:10.1038/nrg2322
- [73] van Rheenen W, Peyrot WJ, Schork AJ, Lee SH, Wray NR. Genetic correlations of polygenic disease traits: from theory to practice. Nat Rev Genet. 2019;20: 567-581. doi:10.1038/s41576-019-0137-z
- [74] Shi H, Mancuso N, Spendlove S, Pasaniuc B. Local Genetic Correlation Gives Insights into the Shared Genetic Architecture of Complex Traits. Am J Hum Genet. 2017;101: 737-751. doi:10.1016/j.ajhg.2017.09.022
- [75] Tanigawa Y, Li J, Justesen JM, Horn H, Aguirre M, DeBoever C, et al. Components of genetic associations across 2,138 phenotypes in the UK Biobank highlight adipocyte biology. Nat Commun. 2019;10: 4064. doi:10.1038/s41467-019-11953-9
- [76] Aguirre M, Tanigawa Y, Venkataraman GR, Tibshirani R, Hastie T, Rivas MA. Polygenic risk modeling with latent trait-related genetic components. Eur J Hum Genet. 2021. doi:10.1038/s41431-021-00813-0
- [77] Burren OS, Reales G, Wong L, Bowes J, Lee JC, Barton A, et al. Genetic feature engineering enables characterisation of shared risk factors in immune-mediated diseases. Genome Med. 2020;12: 106. doi:10.1186/s13073-020-00797-4
- [78] Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A global atlas of genetic associations of 220 deep phenotypes. medRxiv. 2020. doi:10.1101/2020.10.23.20213652
- [79] Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17: 1520-1528. doi:10.1101/gr.6665407
- [80] Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12: 44. doi:10.1186/s13073-020-00742-5
- [81] Choi SW, Mak TS-H, O'Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15: 2759-2772. doi:10.1038/s41596-020-0353-1
- [82] Wray NR, Lin T, Austin J, McGrath JJ, Hickie IB, Murray GK, et al. From Basic Science to Clinical Application of Polygenic Risk Scores: A Primer. JAMA Psychiatry. 2021;78: 101-109. doi:10.1001/jamapsychiatry.2020.3049
- [83] Wand H, Lambert SA, Tamburro C, Iacocca MA, O'Sullivan JW, Sillari C, et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature. 2021;591: 211-219. doi:10.1038/s41586-021-03243-6
- [84] Bernardo R. Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop Sci. 2008;48: 1649-1664. doi:10.2135/cropsci2008.03.0131
- [85] Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet. 2015;97: 576-592. doi:10.1016/j.ajhg.2015.09.001
- [86] Mak TSH, Porsch RM, Choi SW, Zhou X, Sham PC. Polygenic scores via penalized regression on summary statistics. Genet Epidemiol. 2017;41: 469-480. doi:10.1002/gepi.22050
- [87] Zhu X, Stephens M. Bayesian large-scale multiple regression with summary statistics from genome-wide association studies. Ann Appl Stat. 2017;11: 1561-1592. doi:10.1214/17-aoas1046
- [88] Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50: 1219-1224. doi:10.1038/s41588-018-0183-z
- [89] Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10: 1776. doi:10.1038/s41467-019-09718-5
- [90] Lloyd-Jones LR, Zeng J, Sidorenko J, Yengo L, Moser G, Kemper KE, et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat Commun. 2019;10: 5086. doi:10.1038/s41467-019-12653-0
- [91] Qian J, Tanigawa Y, Du W, Aguirre M, Chang C, Tibshirani R, et al. A fast and scalable framework for large-scale and ultrahigh-dimensional sparse regression with application to the UK Biobank. PLoS Genet. 2020;16: e1009141. doi:10.1371/journal.pgen.1009141
- [92] Li R, Chang C, Justesen JM, Tanigawa Y, Qiang J, Hastie T, et al. Fast Lasso method for large-scale and ultrahigh-dimensional Cox model with applications to UK Biobank. Biostatistics. 2020. doi:10.1093/biostatistics/kxaa038
- [93] Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics. 2020. doi:10.1093/bioinformatics/btaa1029
- [94] Li R, Chang C, Tanigawa Y, Narasimhan B, Hastie T, Tibshirani R, et al. Fast Numerical Optimization for Genome Sequencing Data in Population Biobanks. bioRxiv. 2021. doi:10.1101/2021.02.14.431030
- [95] Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11: 3635. doi:10.1038/s41467-020-17374-3
- [96] Mostafavi H, Harpak A, Agarwal I, Conley D, Pritchard JK, Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. Elife. 2020;9. doi:10.7554/eLife.48376
- [97] Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot W, Khera A, et al. Leveraging fine-mapping and non-European training data to improve trans-ethnic polygenic risk scores. medRxiv. 2021. doi:10.1101/2021.01.19.21249483
- [98] Wen X, Stephens M. Using linear predictors to impute allele frequencies from summary or pooled genotype data. Ann Appl Stat. 2010;4: 1158-1182. doi:10.1214/10-aoas338
- [99] Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J, et al. Strong rules for discarding predictors in lasso-type problems. J R Stat Soc Series B Stat Methodol. 2012;74: 245-266. doi:10.1111/j.1467-9868.2011.01004.x
- [100] Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53: 185-194. doi:10.1038/s41588-020-00757-z
- [101] Qian J, Tanigawa Y, Li R, Tibshirani R, Rivas MA, Hastie T. Large-Scale Sparse Regression for Multiple Responses with Applications to UK Biobank. bioRxiv. 2020. doi:10.1101/2020.05.30.125252
- [102] Li R, Tanigawa Y, Justesen JM, Taylor J, Hastie T, Tibshirani R, et al. Survival Analysis on Rare Events Using Group-Regularized Multi-Response Cox Regression. Bioinformatics. 2021. doi:10.1093/bioinformatics/btab095
- [103] Nag A, McCarthy MI, Mahajan A. Large-Scale Analyses Provide No Evidence for Gene-Gene Interactions Influencing Type 2 Diabetes Risk. Diabetes. 2020;69: 2518-2522. doi:10.2337/db20-0224

