Abstract
Several studies have demonstrated the chemopreventive role of ketoconazole in animal models of liver injury. However, the underlying molecular mechanisms of this hepatoprotective effect are poorly understood. The present study assessed the potential of ketoconazole to enhance resistance to carbon tetrachloride-induced hepatotoxicity in vivo in a rat model. Ketoconazole pretreatment adult male rats were intraperitoneally injected with carbon tetrachloride for 24 hr and various hepatic parameters were analyzed. We observed decreased serum transaminases activity, reduced nuclear RelA/p65 expression, and suppressed production of pro-inflammatory cytokines in the liver tissue. Histopathological examination demonstrated ketoconazole pretreatment to extensively prevent liver injury. In addition, it significantly increased nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) protein expression, glutathione (GSH) to oxidized glutathione (GSSG) ratio, and antioxidant enzymes gene expression. These results suggest that ketoconazole pretreatment ameliorates carbon tetrachloride-induced acute liver injury in rats, signifying its anti-inflammatory and antioxidant functions.
INTRODUCTION
The liver is the largest metabolic organ in the human body that is involved in detoxification and excretion of waste material, functions that are fundamental to the survival of any living entity. Liver injury is commonly observed in clinical settings. Carbon tetrachloride (CCl4) is a typical hepatotoxicant, which is widely used to induce acute liver injury or chronic liver fibrosis in experimental rodents, owing to the similarity of its molecular mechanism with humans (Zhang et al., 2017). Many studies have demonstrated that CCl4 is metabolized into a highly reactive trichloromethyl free radical by cytochrome P450 2E1 (CYP2E1) in liver, thereby triggering lipid peroxidation as early as 2 hr after CCl4 intoxication. Therefore, severe inflammatory response and oxidative stress are the hallmarks of CCL4-induced liver injury (Peng et al., 2018; Lee et al., 2015).
Aryl hydrocarbon receptor (AhR) is an evolutionarily old, ligand-activated transcription factor that belongs to the bHLH/PAS (basic helix-loop-helix/Per-Arnt-Sim) family and controls the transcription of a large set of genes. It can be activated by a number of low molecular weight chemicals, ranging from environmental pollutants to dietary chemicals. AhR has long been known to be a cytosolic sensor that is involved in biotransformation and detoxification of small molecules. Currently, it is viewed as a protein with an important functional role in cell and organ development, peripheral and intestinal immunity, and cancer (Esser and Rannug, 2015). It is also involved in regulating the inflammatory response as well as disease tolerance. Generally speaking, sustained or uncontrolled activation of the AhR by tetrachlorodibenzo-p-dioxin (TCDD), coplanar polychlorinated biphenyls (PCBs), and polycyclic aromatic hydrocarbons (PAHs) exposure have been linked to an enhancement of inflammatory signaling, while transient or constitutively low levels of AhR activity may inhibit cytokine production in certain cell types and in vivo models (Beischlag et al., 2008). For example, treatment with β-naphthoflavone, an AhR agonist, repressed the dbcAMP/theophyline-induced astrocytic differentiation of C6 glioma by inhibiting generation of the interleukin 6 (Takanaga et al., 2004). AhR-deficient mice become more sensitive to lipopolysaccharide (LPS) than wild-type mice (Bessede et al., 2014). Moreover, the possible interplay between the AhR and antioxidant role has been explored to a limited extent.
Nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) has been considered as an essential protective factor against oxidative stress; it is known to control adaptive responses to various environmental stressors. Under homeostatic conditions, it is sequestered in the cytosol by binding to kelch-like ECH associating protein 1 (Keap1). During exposure to electrophiles and oxidative toxicity, Nrf2–Keap1 complex is disrupted, and Nrf2 is rapidly transferred to the nucleus, where it binds the antioxidant response elements (AREs) (Nioi et al., 2003; Itoh et al., 2004). The crosstalk between Nrf2 and AhR has been extensively studied. AhR-mediated target gene or metabolic activity could perhaps promote some Nrf2 activation (Miao et al., 2005). Conversely, Nrf2 regulates expression of AhR, subsequently modulating several downstream events of the AhR signaling cascade. For instance, a study reported marked increase in the expression levels of AhR and its downstream genes and proteins in the mouse embryonic fibroblasts from Nrf2+/+ mice than Nrf2−/− mice after TCDD stimulus (Shin et al., 2007). AhR-/- mice and Nrf2-/- mice share some common phenotypes such as increasing sensitivity to infection, carcinogenesis, altered responses to wounding, and metabolic syndrome (Wakabayashi et al., 2010).
Ketoconazole (KCZ) was originally developed and marketed for the treatment of superficial and systemic fungal infections. It has also been used to treat Cushing’s syndrome, female hirsutism, polycystic ovarian syndrome, and male pattern alopecia on an off-label basis. The most adverse effect of oral KCZ is hepatotoxicity, which restricts its clinical application (Gupta and Lyons, 2015). Interestingly, KCZ application could decrease liver enzymes in Cushing’s syndrome (Ollivier et al., 2018). Some studies have also indicated it to relieve the lipopolysaccharide (LPS)-induced liver injury or prevent against hepatocyte injury by CCl4 (Minamiyama et al., 2004; Akhtar et al., 2017). However, the exact mechanism of KCZ-mediated protective effect on liver injury remains unknown. Previous studies confirm that KCZ is an activator of AhR in hepatocytes and owns an ability to activate Nrf2 via AhR signaling in cultured human keratinocytes (Korashy et al., 2007; Tsuji et al., 2012); therefore, we hypothesized that KCZ pretreatment might have protective effects on CCl4-induced acute liver injury in a rat model.
MATERIALS AND METHODS
Animal and environmental conditions
A total of 48 male Sprague-Dawley (SD) rats, weighing 220 ± 20 g, were purchased from the experimental animal center of the Guangzhou University of Chinese Medicine (Animal permit number: SCXK [Yue] 2013-0034). The animals were fed food and water ad libitum under controlled laboratory conditions (temperature, 25 ± 1°C; humidity, 60 ± 10%; and a 12/12 hr light/dark cycle). The rats were acclimated to the laboratory conditions for at least 5 days prior to the experiment. All procedures involving animals were approved and were conducted under the regulations of the Committee on Care and Use of the Guangzhou University of Chinese Medicine, China.
Chemicals and their preparation
Carbon tetrachloride was purchased from Tianjin Fu Yu Fine Chemical Co., Ltd (Tianjin, China). KCZ (1-(4-(4-(((2R,4S)-2-((1H-imidazol-1-yl)methyl)-2-(2,4-dichlorophenyl)-1,3-dioxolan-4-yl)methoxy)phenyl)piperazin-1-yl)ethanone) was procured from the Aladdin Industrial Corporation (Shanghai, China). All other chemicals and reagents were of analytical grade and commercially available. For the experiments, all chemicals were prepared immediately before treatment; CCl4 was dissolved in paraffin oil, whereas KCZ was dispersed in 0.6% sodium carboxymethyl cellulose (CMC-Na). The dosage of CCl4 and KCZ was established according to previous experiments (Yuan et al., 2018; Minamiyama et al., 2004).
Animal treatment
The rats were randomly divided into four groups (12 rats per group) as follows: (I) control: rats were gavaged with vehicle only, (II) model: rats were injected with a single intraperitoneal (i.p.) dose of CCl4-paraffin oil mixture (50% CCl4, v/v, 2 mL/kg body weight), (III) KCZ pretreatment: rats were treated with KCZ (100 mg/kg body weight, p.o.) once a day for 3 days and injected a single i.p. dose of CCl4-paraffin oil mixture (50% CCl4, v/v, 2 mL/kg body weight) 3 hr after the last gavage, and (IV) KCZ only: rats were administered KCZ (100 mg/kg body weight, p.o.) once a day for 3 days. Rats were sacrificed 24 hr after CCl4 challenge under sodium pentothal anesthesia. The blood was collected from abdominal aorta, and a portion of liver tissue specimens were stored at −80°C till further analysis.
Histopathological examination of liver
A part of liver specimen was fixed in 10% formalin solution for 48 hr. It was then embedded in paraffin and sliced into 5-μm-thick sections using the microtome, followed by staining with hematoxylin and eosin (H & E). The tissue sections were then examined under a bright field microscope at different magnifications.
Serum liver enzyme analysis
Serum was obtained by centrifugation at 2000 rpm for 5 min within 1 hr after blood collection. Activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured immediately using an automatic biochemical analyzer and commercially available enzymatic assay kits (Beijing Strong Biotechnologies Inc.; Beijing, China).
Measurement of TNF-α, IL-1β, and IL-6
Concentrations of cytokines, namely tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, were determined using enzyme-linked immunosorbent assay (ELISA) kits (Cusabio Biotech Co., Ltd., Wuhan, China) according to the manufacturer’s instructions. Briefly, frozen liver tissue samples were washed with phosphate-buffered saline (PBS; pH 7.2). Samples were homogenized in a glass homogenizer in PBS (10% w/v), and then kept overnight at −20°C. Homogenized samples were then subjected to freeze–thaw cycles twice to disintegrate the cell membranes, following which the homogenate was centrifuged at 5000 g at 4°C for 5 min. The supernatant was then removed and subjected to ELISA immediately.
Determination of glutathione
The contents of reduced glutathione (GSH) and oxidized glutathione (GSSG) were measured using a commercially available kit according to the manufacturer’s instructions (Beyotime Institute of Biotechnology; Shanghai, China). The absorbance value was monitored at 412 nm on a microplate reader.
RNA isolation and quantification
Total RNA was extracted from liver samples using RNAiso Plus (Takara Bio Inc., Shiga, Japan). Purity of RNA was verified by gel electrophoresis and RNA concentrations were measured spectrophotometrically at 260 nm. Total RNA (2 µg) was reverse-transcribed into first-strand cDNA using PrimeScript RT Master Mix (Takara Bio Inc.). Real-time polymerase chain reaction (PCR) was conducted with SYBR Premix Ex TaqII (Takara Bio Inc.). All procedures were performed according to the manufacturer’s protocol. Relative gene expression was calculated using ΔΔCT method according to a previous study (Pfaffl, 2001). Table 1 lists the sequences of PCR primers used in this study.
Table 1. The sequences of primers used for real-time RT-PCR detection of expression.
| Name |
Primer sequence (5′-3′) |
| AhR |
F:CCATGTCCATGTACCAGTGC |
| R:TGAGCAGCAGTCTGAAGGTG |
| AhRR |
F:ATGGAGATTTTTGTGGGTCACT |
| R:CTCTTTTCTGTGTGCTGTGCTC |
| CYP1A1 |
F: GCCATCTGCTGAGGCTCAAC |
| R: TGAGCAGCAGTCTGAAGGTG |
| GSR |
F: TGAGCCGCCTGAACAACA |
| R: TTGCGTAGCCGTGGATGAC |
| GSTα |
F: AATATGTCCCCCAGACCAAAGA |
| R: GGCAGGCAAGTACCGGTTT |
| NQO1 |
F: GTGAGAAGAGCCCTGATTGT |
| R: CCTGTGATGTCGTTTCTGGA |
| GAPDH |
F: GGCACAGTCAAGGCTGAGAATG |
| R: ATGGTGGTGAAGACGCCAGTA |
Note: AhR: aryl hydrocarbon receptor. AhRR: aryl hydrocarbon receptor repressor; CYP1A1: cytochrome P450 1A1; GSR: Glutathione-disulfide reductase;GSTα:Glutathione S transferase α;NQO1:NAD (P) H-quinone oxidoreductase 1; GAPDH= lyceraldehyde-3- phosphate dehydrogenase.
Hepatic protein isolation and western blot analysis
A frozen liver sample was cut into small pieces and homogenized in a glass homogenizer with commercially available protein extraction kit (KeyGen Biotech Co. Ltd., Nanjing, China). Protein concentrations were determined using the bicinchoninic acid (BCA) method, and a total of 40 μg protein was separated by electrophoresis and blotted onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Cork, Ireland). The membranes were incubated with primary rabbit antibodies against rat HO-1, CYP2E1, Nrf2, Lamin B1, and GAPDH (Proteintech; 1:1,000 dilution) and NF-κB/P65 (CST; 1:1,000 dilution) at 4°C overnight. Then, the membranes were washed thrice and incubated with secondary antibodies (CST; 1:2,000 dilution) at room temperature for 1 hr. ImageJ software was used to measure the intensity of the bands in the membrane.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). One-way analysis of variance and Student–Newman–Keuls multiple comparisons were used to analyze the differences among the groups using the software Stata (version 13.0). A P-value < 0.05 was considered to be statistically significant. Graphs were generated by GraphPad Prism software (version 6.0).
RESULTS
General histopathology examination
As shown in Fig. 1, hepatic histopathological examination in the KCZ-only treatment group revealed no degenerative signs as compared to the normal liver tissues of the vehicle controls. In contrast, CCl4-treated rats demonstrated severe hepatocellular damage, characterized by large areas of necrotic tissue, severe loss of hepatic architecture together with a significant hepatocyte ballooning. Although these findings were also observed in the KCZ pretreatment group, the incidence and severity of histopathological lesions were less prominent in the model animals.
KCZ affects serum enzyme levels
As depicted in Fig. 2, CCl4-challenged rats showed a significant elevation of serum ALT and AST, as compared with the control group (P < 0.01). In contrast, activities of serum AST and ALT in the KCZ pretreatment group reduced considerably as compared with those in the model group (P < 0.01). The KCZ treatment alone caused slight elevations in ALT levels.
KCZ suppresses pro-inflammatory cytokine production
As shown in Fig. 3, CCl4 intoxication caused significant increase in the levels of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, in the liver tissue as compared with that in the control group (P < 0.01). These levels declined in the KCZ pretreatment group as compared with the model group (P < 0.01 or P < 0.05). KCZ treatment alone had no effect on the generation of pro-inflammatory cytokines.
KCZ increases glutathione generation
Fig. 4 demonstrates a mildly elevated hepatic GSH content in the model group as compared with the controls. However, treatment with KCZ resulted in a further elevation of GSH content than that in the KCZ-free rats (P < 0.05). Only oral KCZ caused a mildly higher level of GSSG compared with the control, but CCl4-stimuli resulted in a significantly higher level of GSSG than the control (P < 0.01). Pretreatment with KCZ prevented GSSG production after CCl4 challenge. Similarly, pretreatment with KCZ ameliorated GSH/GSSG ratio in the liver (P < 0.05). The above data suggested that KCZ could induce a mild oxidation stress but alleviate the serious intracellular oxidation status resulting from CCl4 exposure.
KCZ increases AhR, CYP1A1, and AhRR mRNA expression
We used quantitative PCR to examine the effects of KCZ on AhR mRNA expression in rats. As shown in Fig. 5, administration of KCZ increased 2.0-fold expression of AhR mRNA over the control group (P < 0.01), thereby up-regulating the AhRR mRNA levels. In contrast, exposure of CCl4 induced a significant decrease in AhR mRNA levels as compared with the controls (P < 0.05), similarly, both KCZ-treated animals showed an up-regulated expression of the CYP1A1 mRNA as compared with the KCZ-free rats (P < 0.01). The AhRR mRNA expression could be barely detected in the normal group; nevertheless, the expression of AhRR mRNA remarkably increased after KCZ treatment, especially in the KCZ + CCl4-challenged group. No difference was observed between the controls and the models.
KCZ affects antioxidant enzyme gene expression
To further determine whether expression of AhR–Nrf2-related genes was affected, a quantitative RT-PCR analysis was performed that revealed mRNAs of glutathione-disulfide reductase (GSR), glutathione S transferase α (GSTα), and NAD(P)H-quinone oxidoreductase 1 (NQO1). As shown in Fig. 6, GSTα gene was found to be down-regulated 5-fold after CCl4 challenge (P < 0.01), whereas pretreatment with KCZ reversed this down-regulation induced by CCl4. A single dose of CCl4 challenge failed to change the mRNA expression of GSR and NQO1; however, KCZ-only administration significantly increased the levels of GSR and NQO1 mRNAs as compared with the control group (P < 0.05 or P < 0.01). Administration of KCZ in combination with CCl4 challenge also resulted in a slight elevation versus the control or model group (P < 0.05).
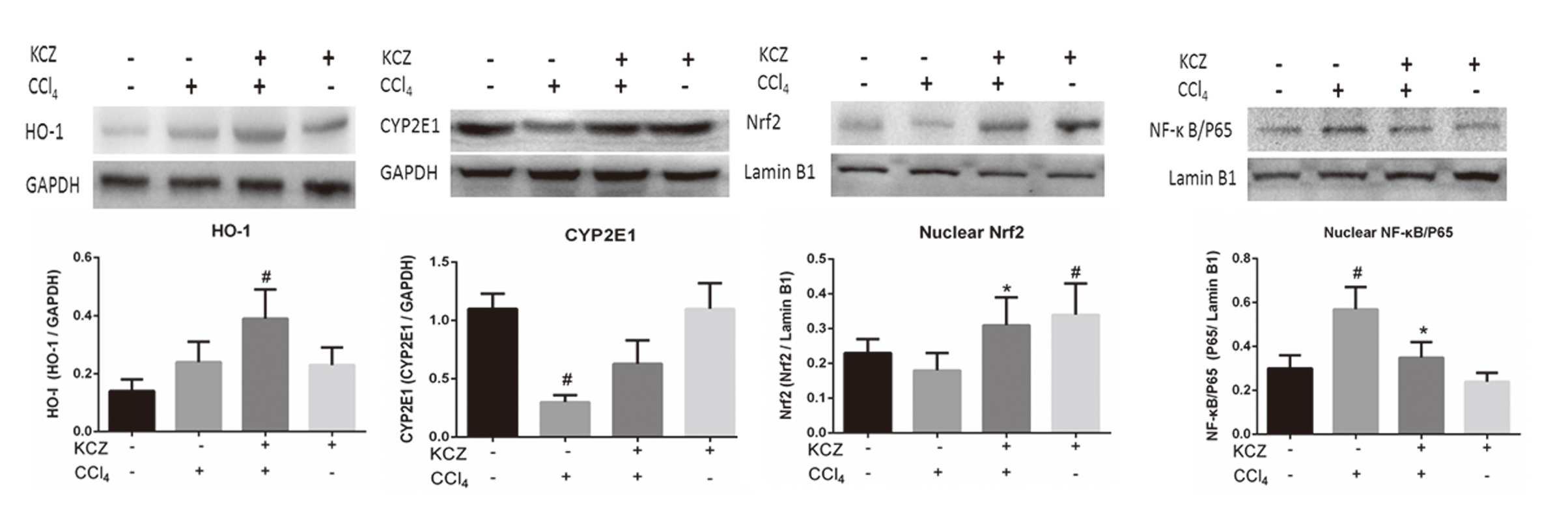
KCZ affects protein levels of HO-1, Nrf2, and NF-κB
We next examined the protein levels of heme oxygenase (HO)-1, CYP2E1, Nrf2, and NF-Κb/p65 by western blotting to determine whether KCZ could induce the Nrf2/ARE pathway during CCl4-induced liver injury. As shown in Fig. 7, there were no significant differences among the control, model, and KCZ-only treatment groups in HO-1 protein levels; however, pretreatment with KCZ following CCl4-intoxication augmented the expression levels of HO-1 (P < 0.05). Both KCZ-treated animals showed a higher level of Nrf2 protein in the nuclear than those in the KCZ-free rats (P < 0.05). Elevated levels of nuclear NF-κB/p65 protein were observed in the model group than other groups (P < 0.05). CCl4-intoxication obviously suppressed CYP2E1 expression (P < 0.05); however, pretreatment with KCZ relieved CYP2E1 suppression after CCl4 challenge.
DISCUSSION
Ketoconazole is an anti-fungal agent that was approved as a drug against a broad range of fungal infections in 1981; however, severe hepatic side-effects associated with it led to its restricted use (Maertens, 2004; Gupta and Lyons, 2015). Several lines of evidence indicate that KCZ could transform into a toxic metabolite, N-deacetyl-ketoconazole (DAK), which had a greater intrinsic toxicity than the original compound (Rodriguez et al., 1999; Rodriguez and Acosta, 1997). In our current study, we found that KCZ treatment alone could cause a slight increase in the ALT and GSSG levels; however, pretreatment with KCZ ameliorated CCl4-induced liver injury in rats, suggesting preconditioning to be one of the mechanisms. Preconditioning (also named adaptive response) is a phenomenon in which a prior exposure to an appropriate low dose of a toxic agent or stress could resist the toxic effects exerted by a subsequent harmful exposure of the same, a related, or an unrelated toxin/stress (Calabrese, 2016). It was reported that a prior low dose of CCl4 induced a state of refractoriness to a subsequent much higher dose of CCl4 (Ugazio et al., 1972). So far, LPS is one of the most frequently reported conditioning agents. For instance, pretreatment with nonlethal LPS dose has been shown to exert protective effects, manifested by attenuation of tissue injury and prolonged survival (Szabó et al., 1994; Bautista and Spitzer, 1995). A recent research found that exposure to LPS activated AhR, consequently leading to a down-regulation of early inflammatory gene expression, which, in turn, protected the mice against subsequent stimuli. Another study demonstrated bacterial pigments that serve as virulence factors, activate the host defense mechanism against acute and chronic bacterial infections upon binding to AhR (Moura-Alves et al., 2014). In AhR knock-out mice, apoptotic cells induce transcription of inflammatory cytokines genes in the macrophages leading to the development of autoimmune diseases. Apoptotic cells activate AhR in mouse macrophages and dendritic cells by the generation of immunomodulatory cytokine IL-10, which blocks inflammatory response and maintains normal physiological homeostasis (Shinde et al., 2018). These findings demonstrated the role of AhR in contributing to adaptive response. Ketoconazole has been identified as an activator of AhR (Korashy et al., 2007; Novotna et al., 2014). It is therefore possible that the protective effect of pretreatment with KCZ against CCl4-induced liver injury in rats may involve AhR signaling.
CYP1A1 is considered to be a prototypical target gene of AhR. Our study revealed KCZ treatment to enhance CYP1A1 mRNA expression, which is consistent with previous studies (Korashy et al., 2007; Novotna et al., 2014). AhR-mediated adaptive response may involve the inhibition of NF-κB, which contributes to the production of inflammatory cytokines. Research confirmed that activation of AhR signaling pathway could promote proteasome-mediated degradation of RelA/p65 protein following its ubiquitination, thus inhibiting the production of inflammatory cytokines (Domínguez-Acosta et al., 2018). Apart from the canonical NF-κB pathway, AhR can regulate the expression of RelB, a component of the alternative pathway, which diminishes the inflammatory reaction (Baglole et al., 2008). CCl4 exposure affects various intracellular signaling pathways. For instance, it activates the NF-κB signaling pathway resulting in a release of various pro-inflammatory mediators (Yamada and Fausto, 1998; Kiso et al., 2012). In the present study, we found that KCZ pretreatment reduced the generation of pro-inflammatory cytokines following CCl4 intoxication in the liver tissue, which might link the mechanism that KCZ activated AhR pathway and decreased nuclear levels of p65 protein. In addition to the role for AhR in controlling the extent of inflammatory reactions, the cross talk between the antioxidant response pathway controlled by Nrf2 and the xenobiotic response pathway controlled by AhR has been studied extensively. We investigated the effect of KCZ on suppressing oxidative stress in these models.
There are two possible mechanisms of AhR-mediated activation of Nrf2 have been suggested: (1) reactive oxygen species (ROS) production caused by CYP1A1 metabolism might facilitate Nrf2 nuclear translocation (Barouki and Morel, 2001), and (2) When AhR is activated, it transfers to the nucleus and dimerizes with AhR nuclear translocator (ARNT), then the AhR-ARNT complex activates the xenobiotic-responsive element (XRE) transcription. DNA sequence analyses reveal that there are XRE-like elements in the mouse Nrf2 promoter, which directly up-regulate Nrf2 mRNA and protein expressions (Miao et al., 2005). Our study revealed KCZ-mediated translocation of Nrf2 to the nucleus, the mechanism of which may involve AhR activation. NQO1 and GSR are prototypical Nrf2 targets that protect against several highly reactive and potentially damaging endogenous and exogenous molecules by catalyzing their two-electron reductive metabolism and detoxification (Ross, 2004). Similarly, GSTs are a superfamily of multifunctional xenobiotic metabolizing enzymes that detoxify the foreign molecules by conjugation of GSH (Oakley, 2011). GSH concentration, in turn, is maintained by GSR (Couto et al., 2016). In the present study, the mRNA expression of GSR, GSTα, and NQO1 was moderately up-regulated in response to KCZ treatment. Another molecule with antioxidant activity is HO-1, which catalyzes the first and rate-limiting step in the catabolism of pro-oxidant heme into biliverdin, which can be reduced to the antioxidant bilirubin by biliverdin reductase to unleash antioxidant and anti-inflammatory effects (Wunder and Potter, 2003). Accordingly, we observed a high level of HO-1 protein in pretreatment with KCZ following CCl4-intoxication rats. Reduced GSH is a major endogenous antioxidant produced by cells, whereas its oxidized form GSSG is indicative of oxidative stress. As a marker for the intracellular redox equilibrium, we employed GSH/GSSG ratio to evaluate the oxidative stress. We found that KCZ-pretreated rats could effectively inhibit the decrease in the GSH/GSSG ratio induced by CCl4 intoxication.
Ketoconazole is a widely accepted inhibitor of p450, and CCl4 is metabolized by CYP2E1 in the liver into toxic free radicals. Some relevant studies have explored the effects of ketoconazole on CYP2E1 enzyme activity. Rats were given KCZ (140, 280, 420 μmol/kg·d-1) for 7 days had no significant inhibitory effect on activity of CYP2E1 in the liver (Cao et al., 2007). Another study revealed that KCZ did not markedly inhibit CYP2E1 activity at low concentration but could inhibit CYP2E1-catalyzed activity at high concentration in human liver microsomes (Zhang et al., 2002). Recent study also demonstrated KCZ had no inhibition effect on CYP2E1 activity in human liver microsomes (Krasulova et al., 2016). It seems that KCZ may be not specific for CYP2E1 inhibitor. In our study, we did not find KCZ to inhibit CYP2E1 protein expression, but we found pretreatment with KCZ alleviated the inhibitory effect of CCl4 on CYP2E1 protein levels. We also found CCl4 challenge resulted in down-regulation of AhR mRNA, whereas KCZ treatment up-regulated AhR mRNA expression. The mechanism remains unclear; one possible mechanism is the autoregulation of AHR expression by its own agonists. However, this is not a definite explanation (Harper et al., 2006). Anyway, alterations in AhR expression may have physiological and toxicological consequences (Englert et al., 2012).
It seems that activation of AhR–Nrf2 pathway promotes the host resistance to subsequent harmful exposure; however, this was not the case. Notably, Nrf2 activation follows a biphasic dose–response relationship—a protective effect at low dose is followed by adverse responses at high doses. TCDD, a potent agonist to AhR, was reported to exacerbate liver damage when administered to mice 48 hr before CCl4 challenge (Mejia-Garcia et al., 2013); this reinforces the notion that adaptive response is based on several key factors related to the characteristics of the conditioning agent and dosage, together with protective windows. Normally, a certain level of Nrf2 activation in healthy cells is beneficial in maintaining homeostasis; however, cancer cells appear to hijack Nrf2-mediated antioxidant to deal with various stresses, such as metabolic stress, mitotic stress, and oxidative stress, which promote the tumorigenic state (Kensler and Wakabayashi, 2010).
In conclusion, we first demonstrated the protective effect of KCZ, which ameliorated CCl4-induced liver injury in an animal model. A possible mechanism of this effect could be activation of coupled AhR and Nrf2 gene batteries. Although KCZ lacks potential to develop into a hepatoprotective agent owing to its well-known hepatotoxicity, AhR–Nrf2 might be considered as a promising new target for prevention of acute liver injury. The results of the present study also expand our insights into the pharmacological activity of KCZ. KCZ could serve as a promising medicinal tool to study the functions of AhR–Nrf2 pathway. In addition, our findings may pave the way for a better understanding of the mechanisms of some hepatoprotective agents.
ACKNOWLEDGMENT
This work was supported by a grant from Science and Technology Plan Projects of Guangdong Province (2013B090600020), and major Research Projects of First-class Disciplines in Guangzhou University of Traditional Chinese Medicine (33).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Akhtar, U., Ahmad, M., Tayyeb, A. and Ali, G. (2017): Comparative study of cytochrome p450 inhibitors on cultured mouse hepatocytes. Pharmacologyonline, 1, 68-88.
- Baglole, C.J., Maggirwar, S.B., Gasiewicz, T.A., Thatcher, T.H., Phipps, R.P. and Sime, P.J. (2008): The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J. Biol. Chem., 283, 28944-28957.
- Barouki, R. and Morel, Y. (2001): Repression of cytochrome P450 1A1 gene expression by oxidative stress: mechanisms and biological implications. Biochem. Pharmacol., 61, 511-516.
- Bautista, A.P. and Spitzer, J.J. (1995): Acute endotoxin tolerance downregulates superoxide anion release by the perfused liver and isolated hepatic nonparenchymal cells. Hepatology, 21, 855-862.
- Beischlag, T.V., Luis Morales, J., Hollingshead, B.D. and Perdew, G.H. (2008): The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr., 18, 207-250.
- Bessede, A., Gargaro, M., Pallotta, M.T., Matino, D., Servillo, G., Brunacci, C., Bicciato, S., Mazza, E.M., Macchiarulo, A., Vacca, C., Iannitti, R., Tissi, L., Volpi, C., Belladonna, M.L., Orabona, C., Bianchi, R., Lanz, T.V., Platten, M., Della Fazia, M.A., Piobbico, D., Zelante, T., Funakoshi, H., Nakamura, T., Gilot, D., Denison, M.S., Guillemin, G.J., DuHadaway, J.B., Prendergast, G.C., Metz, R., Geffard, M., Boon, L., Pirro, M., Iorio, A., Veyret, B., Romani, L., Grohmann, U., Fallarino, F. and Puccetti, P. (2014): Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature, 511, 184-190.
- Calabrese, E.J. (2016): Preconditioning is hormesis part II: How the conditioning dose mediates protection: Dose optimization within temporal and mechanistic frameworks. Pharmacol. Res., 110, 265-275.
- Cao, A., Shi, C., Liu, Y. and Liao, M. (2007): Effects of ketoconazole on rat liver cytochrome P450. Chin. J. New Drug., 16, 285-287.
- Couto, N., Wood, J. and Barber, J. (2016): The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med., 95, 27-42.
- Domínguez-Acosta, O., Vega, L., Estrada-Muñiz, E., Rodríguez, M.S., Gonzalez, F.J. and Elizondo, G. (2018): Activation of aryl hydrocarbon receptor regulates the LPS/IFNγ-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65. Biochem. Pharmacol., 155, 141-149.
- Englert, N.A., Turesky, R.J., Han, W., Bessette, E.E., Spivack, S.D., Caggana, M., Spink, D.C. and Spink, B.C. (2012): Genetic and epigenetic regulation of AHR gene expression in MCF-7 breast cancer cells: role of the proximal promoter GC-rich region. Biochem. Pharmacol., 84, 722-735.
- Esser, C. and Rannug, A. (2015): The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev., 67, 259-279.
- Gupta, A.K. and Lyons, D.C. (2015): The rise and fall of oral ketoconazole. J. Cutan. Med. Surg., 19, 352-357.
- Harper, P.A., Riddick, D.S. and Okey, A.B. (2006): Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem. Pharmacol., 72, 267-279.
- Itoh, K., Tong, K.I. and Yamamoto, M. (2004): Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med., 36, 1208-1213.
- Kensler, T.W. and Wakabayashi, N. (2010): Nrf2: friend or foe for chemoprevention? Carcinogenesis, 31, 90-99.
- Kiso, K., Ueno, S., Fukuda, M., Ichi, I., Kobayashi, K., Sakai, T., Fukui, K. and Kojo, S. (2012): The role of Kupffer cells in carbon tetrachloride intoxication in mice. Biol. Pharm. Bull., 35, 980-983.
- Korashy, H.M., Shayeganpour, A., Brocks, D.R. and El-Kadi, A.O. (2007): Induction of cytochrome P450 1A1 by ketoconazole and itraconazole but not fluconazole in murine and human hepatoma cell lines. Toxicol. Sci., 97, 32-43.
- Krasulova, K., Siller, M., Holas, O., Dvorak, Z. and Anzenbacher, P. (2016): Enantiospecific effects of chiral drugs on cytochrome P450 inhibition in vitro. Xenobiotica, 46, 315-324.
- Lee, I.-C., Kim, S.-H., Baek, H.-S., Moon, C., Kim, S.H., Kim, Y.B., Yun, W.K., Kim, H.C. and Kim, J.C. (2015): Protective effects of diallyl disulfide on carbon tetrachloride-induced hepatotoxicity through activation of Nrf2. Environ. Toxicol., 30, 538-548.
- Maertens, J.A. (2004): History of the development of azole derivatives. Clin. Microbiol. Infect., 10 (Suppl 1), 1-10.
- Mejia-Garcia, A., Sanchez-Ocampo, E.M., Galindo-Gomez, S., Shibayama, M., Reyes-Hernandez, O., Guzman-Leon, S., Gonzalez, F.J. and Elizondo, G. (2013): 2,3,7,8-Tetrachlorodibenzo-p-dioxin enhances CCl4-induced hepatotoxicity in an aryl hydrocarbon receptor-dependent manner. Xenobiotica, 43, 161-168.
- Miao, W., Hu, L., Scrivens, P.J. and Batist, G. (2005): Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem., 280, 20340-20348.
- Minamiyama, Y., Takemura, S., Toyokuni, S., Imaoka, S., Funae, Y., Hirohashi, K., Yoshikawa, T. and Okada, S. (2004): CYP3A induction aggravates endotoxemic liver injury via reactive oxygen species in male rats. Free Radic. Biol. Med., 37, 703-712.
- Moura-Alves, P., Faé, K., Houthuys, E., Dorhoi, A., Kreuchwig, A., Furkert, J., Barison, N., Diehl, A., Munder, A., Constant, P., Skrahina, T., Guhlich-Bornhof, U., Klemm, M., Koehler, A.B., Bandermann, S., Goosmann, C., Mollenkopf, H.J., Hurwitz, R., Brinkmann, V., Fillatreau, S., Daffe, M., Tümmler, B., Kolbe, M., Oschkinat, H., Krause, G. and Kaufmann, S.H. (2014): AhR sensing of bacterial pigments regulates antibacterial defence. Nature, 512, 387-392.
- Nioi, P., McMahon, M., Itoh, K., Yamamoto, M. and Hayes, J.D. (2003): Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem. J., 374, 337-348.
- Novotna, A., Korhonova, M., Bartonkova, I., Soshilov, A.A., Denison, M.S., Bogdanova, K., Kolar, M., Bednar, P. and Dvorak, Z. (2014): Enantiospecific effects of ketoconazole on aryl hydrocarbon receptor. PLoS One, 9, e101832.
- Oakley, A. (2011): Glutathione transferases: a structural perspective. Drug Metab. Rev., 43, 138-151.
- Ollivier, M., Haissaguerre, M., Ferriere, A. and Tabarin, A. (2018): Should we avoid using ketoconazole in patients with severe Cushing’s syndrome and increased levels of liver enzymes? Eur. J. Endocrinol., 179, L1-L2.
- Peng, X., Dai, C., Liu, Q., Li, J. and Qiu, J. (2018): Curcumin attenuates on carbon tetrachloride-induced acute liver injury in mice via modulation of the Nrf2/HO-1 and TGF-β1/Smad3 pathway. Molecules, 23, 215.
- Pfaffl, M.W. (2001): A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res., 29, e45.
- Rodriguez, R.J. and Acosta, D. Jr. (1997): N-deacetyl ketoconazole-induced hepatotoxicity in a primary culture system of rat hepatocytes. Toxicology, 117, 123-131.
- Rodriguez, R.J., Proteau, P.J., Marquez, B.L., Hetherington, C.L., Buckholz, C.J. and O’Connell, K.L. (1999): Flavin-containing monooxygenase-mediated metabolism of N-deacetyl ketoconazole by rat hepatic microsomes. Drug Metab. Dispos., 27, 880-886.
- Ross, D. (2004): Quinone reductases multitasking in the metabolic world. Drug Metab. Rev., 36, 639-654.
- Shin, S., Wakabayashi, N., Misra, V., Biswal, S., Lee, G.H., Agoston, E.S., Yamamoto, M. and Kensler, T.W. (2007): NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol. Cell. Biol., 27, 7188-7197.
- Shinde, R., Hezaveh, K., Halaby, M.J., Kloetgen, A., Chakravarthy, A., da Silva Medina, T., Deol, R., Manion, K.P., Baglaenko, Y., Eldh, M., Lamorte, S., Wallace, D., Chodisetti, S.B., Ravishankar, B., Liu, H., Chaudhary, K., Munn, D.H., Tsirigos, A., Madaio, M., Gabrielsson, S., Touma, Z., Wither, J., De Carvalho, D.D. and McGaha, T.L. (2018): Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol., 19, 571-582.
- Szabó, C., Thiemermann, C., Wu, C.C., Perretti, M. and Vane, J.R. (1994): Attenuation of the induction of nitric oxide synthase by endogenous glucocorticoids accounts for endotoxin tolerance in vivo. Proc. Natl. Acad. Sci. USA, 91, 271-275.
- Takanaga, H., Yoshitake, T., Yatabe, E., Hara, S. and Kunimoto, M. (2004): Beta-naphthoflavone disturbs astrocytic differentiation of C6 glioma cells by inhibiting autocrine interleukin-6. J. Neurochem., 90, 750-757.
- Tsuji, G., Takahara, M., Uchi, H., Matsuda, T., Chiba, T., Takeuchi, S., Yasukawa, F., Moroi, Y. and Furue, M. (2012): Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: the basis of its anti-inflammatory effect. J. Invest. Dermatol., 132, 59-68.
- Ugazio, G., Koch, R.R. and Recknagel, R.O. (1972): Mechanism of protection against carbon tetrachloride by prior carbon tetrachloride administration. Exp. Mol. Pathol., 16, 281-285.
- Wakabayashi, N., Slocum, S.L., Skoko, J.J., Shin, S. and Kensler, T.W. (2010): When NRF2 talks, who’s listening? Antioxid. Redox Signal., 13, 1649-1663.
- Wunder, C. and Potter, R.F. (2003): The heme oxygenase system: its role in liver inflammation. Curr. Drug Targets Cardiovasc. Haematol. Disord., 3, 199-208.
- Yamada, Y. and Fausto, N. (1998): Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am. J. Pathol., 152, 1577-1589.
- Yuan, Z.-W., Li, Y.-Z., Liu, Z.-Q., Feng, S.L., Zhou, H., Liu, C.X., Liu, L. and Xie, Y. (2018): Role of tangeretin as a potential bioavailability enhancer for silybin: pharmacokinetic and pharmacological studies. Pharmacol. Res., 128, 153-166.
- Zhang, D.-G., Zhang, C., Wang, J.-X., Wang, B.W., Wang, H., Zhang, Z.H., Chen, Y.H., Lu, Y., Tao, L., Wang, J.Q., Chen, X. and Xu, D.X. (2017): Obeticholic acid protects against carbon tetrachloride-induced acute liver injury and inflammation. Toxicol. Appl. Pharmacol., 314, 39-47.
- Zhang, W., Ramamoorthy, Y., Kilicarslan, T., Nolte, H., Tyndale, R.F. and Sellers, E.M. (2002): Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos., 30, 314-318.