Letter
Methylmercury-induced brain neuronal death in CHOP-knockout mice
2024 Volume 49 Issue 2 Pages 55-60
Details
2024 Volume 49 Issue 2 Pages 55-60
Apoptosis is one of the hallmarks of MeHg-induced neuronal cell death; however, its molecular mechanism remains unclear. We previously reported that MeHg exposure induces neuron-specific ER stress in the mouse brain. Excessive ER stress contributes to apoptosis, and CHOP induction is considered to be one of the major mechanisms. CHOP is also increased by MeHg exposure in the mouse brain, suggesting that it correlates with increased apoptosis. In this study, to clarify whether CHOP mediates MeHg-induced apoptosis, we examined the effect of CHOP deletion on MeHg exposure in CHOP-knockout mice. Our data showed that CHOP deletion had no effect on MeHg exposure-induced weight loss or hindlimb impairment in mice, nor did it increase apoptosis or inhibit neuronal cell loss. Hence, CHOP plays little role in MeHg toxicity, and other apoptotic pathways coupled with ER stress may be involved in MeHg-induced cell death.
MeHg, the causative agent of Minamata disease, is known to induce neuronal apoptosis (Kunimoto, 1994; Nagashima et al., 1996; Fujimura et al., 2009a). In general, apoptotic signaling pathways are mediated by cell surface death receptors, DNA damage, and mitochondrial and endoplasmic reticulum (ER) stress. We have previously demonstrated that the chaperone activity of protein disulfide isomerase is reduced by S-merculation, which induces ER stress, leading to cytotoxicity (Makino et al., 2015). In addition, in vivo analysis has revealed that ER stress is induced specifically in neurons in brain regions damaged by MeHg (Hiraoka et al., 2021). Thus, ER stress may contribute to MeHg-induced apoptosis in neurons; however, the exact molecular mechanism that induces apoptosis is unknown.
Under ER stress, cells maintain protein homeostasis via the unfolded protein response (UPR), which consists of transcriptional regulation mediated by three distinct ER stress sensor proteins localized to the ER membrane: inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Walter and Ron, 2011). In vitro studies have shown that MeHg activates PERK and ATF6, respectively (Hiraoka et al., 2017) and induces the expression of C/EBP homologous protein (CHOP), a downstream apoptosis-promoting factor (Makino et al., 2015). Furthermore, the PERK-CHOP pathway is activated in the brains of MeHg-exposed mice and correlates with increased apoptosis (Nomura et al., 2022). Therefore, CHOP might mediate MeHg-induced apoptosis.
CHOP is expressed at very low levels under physiological conditions but is strongly transcriptionally induced in response to ER stress. CHOP is a transcription factor that regulates the expression of various anti-apoptotic and pro-apoptotic factors, and the induction of CHOP is considered one of the major pathways of ER stress-induced apoptosis (Oyadomari and Mori, 2004; Nishitoh, 2012). Several studies have suggested that CHOP is involved in neuronal cell death in brain disorders. For example, CHOP deficiency in mice attenuated ischemia-induced neuronal death caused by bilateral carotid occlusion (Tajiri et al., 2004). CHOP-knockout mice were resistant to apoptosis of substantia nigra dopaminergic neurons induced by the neurotoxin 6-hydroxydopamine (6-OHDA) (Silva et al., 2005). On the other hand, the exact role of CHOP in MeHg-induced neuronal death remains unclear.
It is important to understand the processes and molecules that induce apoptosis to prevent MeHg neurotoxicity. In this study, we examined the protective role of CHOP deletion against MeHg neurotoxicity in CHOP-knockout mice.
Wild-type C57BL6N/Jc1 mice were purchased from CLEA Japan (Tokyo, Japan). Mice lacking the CHOP gene (CHOP-knockout mice; C57BL/6 background) were provided by Dr. Seiichi Oyadomari (Oyadomari et al., 2002). CHOP-knockout mice show normal growth, development, and fertility. All mice were housed in plastic cages (three animals per cage) and allowed free access to food (CE-2; CLEA Japan) and water. The animal facility was maintained at 25 ± 2°C with a relative humidity of 65% ± 5% under a 12-hr light/dark cycle. Mice were euthanized by cardiac blood sampling under deep anesthesia with isoflurane and transcardially perfused with saline. All procedures involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institute for Minamata Disease and were approved by the Animal Ethics and Management Committee of the National Institute for Minamata Disease (No. 050315-1).
MeHg administrationTwo-month-old male mice were randomly divided into two groups: a vehicle group and an MeHg-exposed group. MeHg (Tokyo Chemical Industry, Tokyo, Japan) was dissolved at 30 ppm in distilled water containing equimolar amounts of glutathione (GSH) (Fujifilm Wako Pure Chemical, Osaka, Japan) and administered via drinking water for 8 weeks, as previously described (Fujimura et al., 2009b). The vehicle group received drinking water containing GSH only.
Measurement of mercury contentThe total concentration of mercury (Hg) in the cerebrum and blood was determined by the oxygen combustion–gold amalgamation method using an MA-2 mercury analyzer with the MA2000 software version 1.7.8. (Nippon Instruments, Tokyo, Japan), as previously described (Fujimura et al., 2009b; Hiraoka et al., 2021).
Hindlimb extension testHindlimb impairment is a neurological symptom in rodent models of MeHg poisoning (Fujimura and Usuki, 2020; Nomura et al., 2022). To evaluate hindlimb impairment caused by MeHg poisoning, the mice were suspended in their tail, and the extent of hindlimb extension was observed for 15 sec. If both hindlimbs spread widely outward from the abdomen, a score of 0 was assigned (normal phenotype). If one hindlimb was retracted or both hindlimbs were partially retracted toward the abdomen without touching it, a score of −1 was assigned (mild defect). If both hindlimbs were partially retracted toward the abdomen without touching each other, a score of −2 was assigned (moderate defect). If both hindlimbs were fully clasped toward the abdomen and touched each other, a score of −3 was assigned (severe defect).
Histological analysisThe left brain was fixed with 4% paraformaldehyde in 0.1 M phosphate buffer immediately after dissection. After embedding the brain in paraffin, serial sagittal sections of 5-μm thickness were prepared using a microtome and mounted on glass slides. Brain sections were deparaffinized in xylene and rehydrated in a graded series of ethanol solutions before staining.
Apoptotic cells were detected by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay using an In situ Cell Death Detection Kit, TMR red (#12156792910; Roche, Basel, Switzerland), according to the manufacturer’s instructions. The sections were mounted in VECTASHIELD Vibrance Antifade Mounting Medium with DAPI (Vector Laboratories, Newark, NJ, USA). Images were captured and analyzed using an ECLIPSE Ti confocal microscope with NIS-Elements AR imaging software version 4.00.06 (Nikon Instruments, Tokyo, Japan).
Immunostaining was performed using the VECTASTAIN Elite ABC Kit (Vector Laboratories) according to the manufacturer’s instructions with a rabbit anti-neuronal nuclei (NeuN) antibody (#GTX133127, 1:250; GeneTex, Irvine, CA, USA) according to the manufacturer’s instructions. Deparaffinized sections were boiled for 20 min in 10 mM citrate buffer at pH 6 (Genemed Biotechnologies, South San Francisco, CA, USA) for antigen retrieval, and endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 20 min at room temperature before immunostaining. After the antigen–antibody reaction, the sections were developed using a DAB Substrate Kit (Vector Laboratories) according to the manufacturer’s instructions and mounted using Eukitt mounting media (ORSAtec, Bobingen, Germany). Images were captured and analyzed using a BX50 microscope (Evident, Tokyo, Japan) with FLOVEL image filling system version 2.30.03 (Flovel, Tokyo, Japan)
Statistical analysisQuantitative data are presented as the means ± standard error of the mean (S.E.). Statistical analyses were performed using the GraphPad Prism software version 10.0.2 (GraphPad Software, San Diego, CA, USA). Differences between two means were analyzed using two-way analysis of variance (ANOVA) followed by an uncorrected Fisher's LSD test and Tukey’s post hoc test. All p-values of < 0.05 were considered statistically significant.
Neuropathological changes such as neuronal loss and increased apoptotic cells in the cerebral cortex were observed in mice treated with drinking water containing 30 ppm MeHg for 8 weeks (Fujimura et al., 2009b). To clarify the role of CHOP in the neurotoxicity of MeHg, wild-type and CHOP-knockout mice were exposed to MeHg using the same method as in a previous study (Fig. 1A). First, we analyzed the total Hg content in the blood and cerebrum to determine the effect of CHOP deletion on MeHg accumulation. In wild-type mice, Hg was significantly increased in these tissues. On the other hand, the same level of Hg accumulated in CHOP-knockout mice, confirming that there was no difference between the two (Fig. 1B). Next, we focused on weakening the hindlimb extension reflex as a neurological symptom of MeHg intoxication in rodents and examined it in wild-type and CHOP-knockout mice. However, no differences in this symptom were observed (Fig. 2A). There was no difference in body weight between the wild-type and CHOP-knockout mice throughout the analysis period (Fig. 2B).
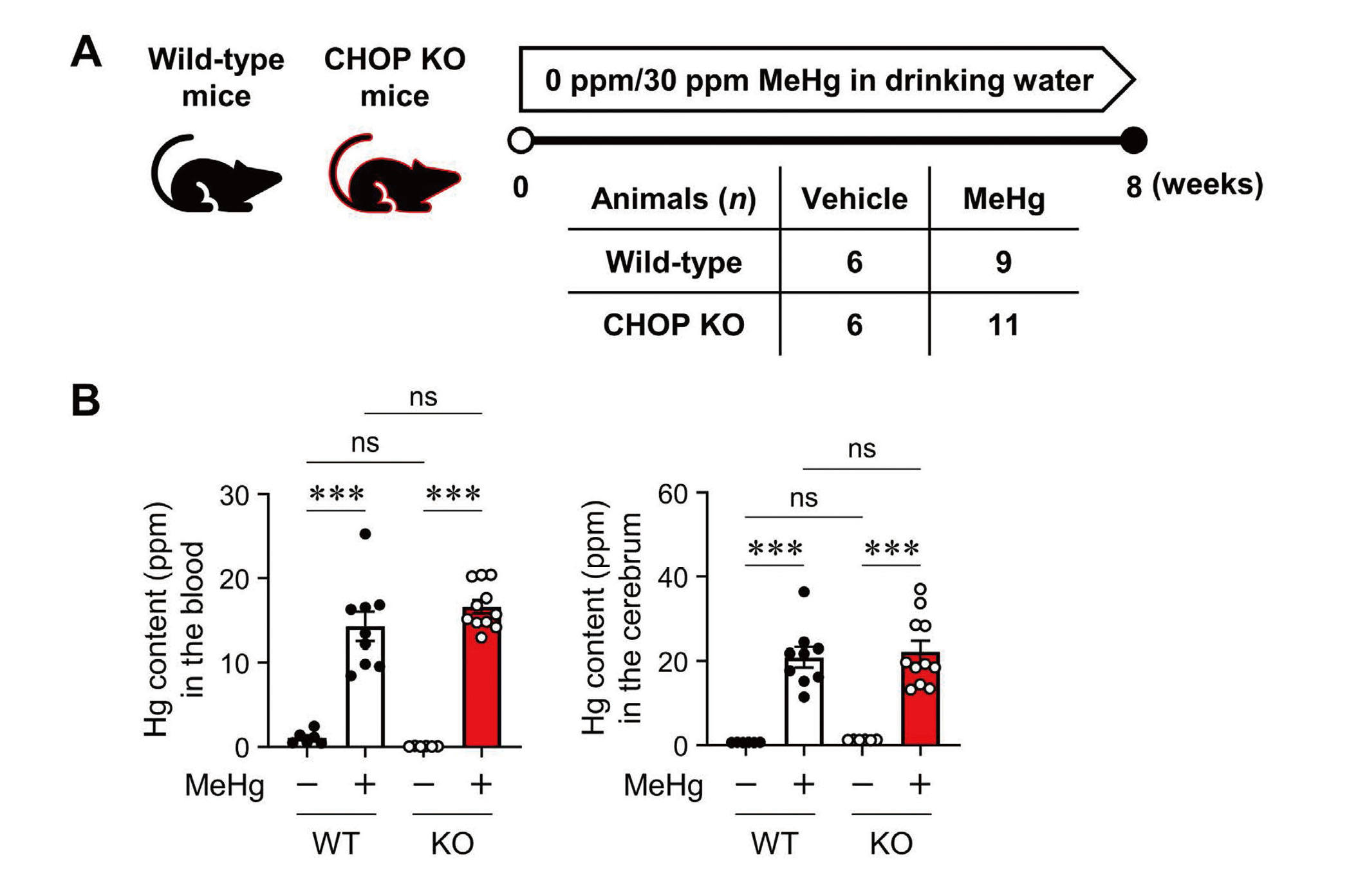
Mercury (Hg) accumulation in CHOP-knockout mice exposed to methylmercury (MeHg). (A) Schematic representation of subchronic exposure to MeHg. Wild-type (WT) and CHOP-knockout (KO) mice were administered either 0 ppm (vehicle) or 30 ppm MeHg. After 8 weeks of MeHg exposure, the mice were sacrificed for further analysis, as shown in Figs. 1b and 3 (WT + Vehicle, n = 6; WT + MeHg, n = 9; KO + Vehicle, n = 6; KO + MeHg, n = 11). (B) Quantification of the total Hg concentration in the blood (left) and cerebrum (right). Data are presented as the mean ± S.E. (n = 6-11; ***p < 0.001 by two-way ANOVA followed by uncorrected Fisher's LSD test; ns, not significant).
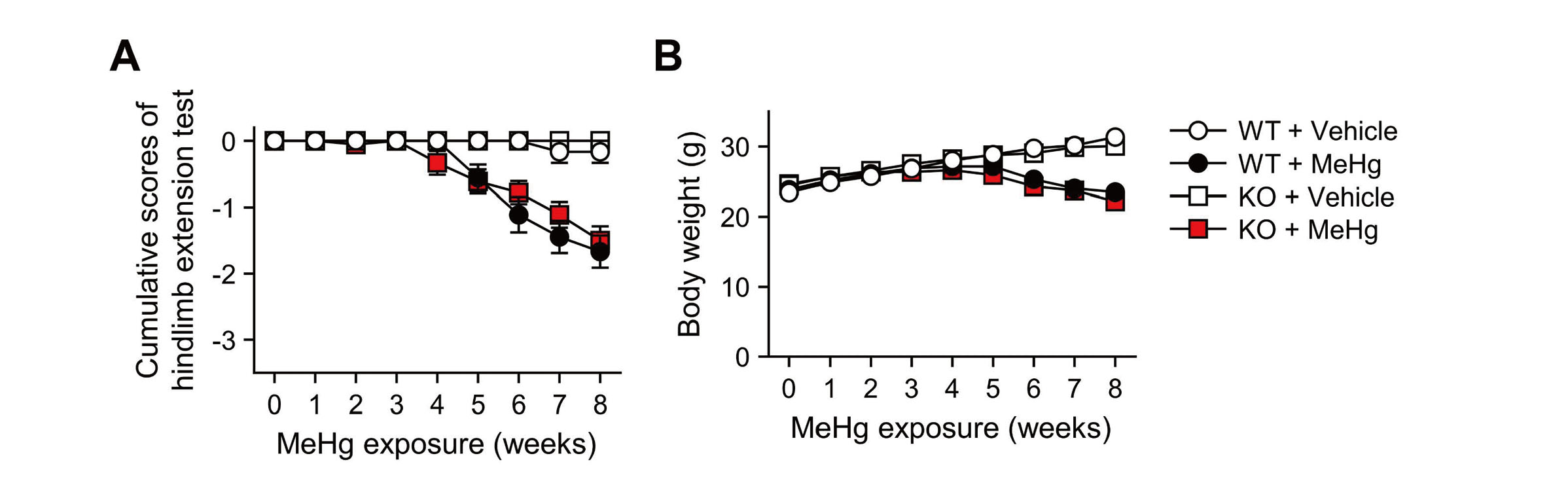
Characteristics of CHOP-knockout mice exposed to methylmercury (MeHg). (A) Hindlimb extension scores and (B) body weight of wild-type (WT) and CHOP-knockout (KO) mice. These mice received either 0 ppm (vehicle) or 30 ppm MeHg via drinking water for 8 weeks. Mice that withdrew because of death were plotted with the last measurement throughout the rest of the analysis. Data are presented as the mean ± S.E. (n = 6-18; No significant differences between WT and KO groups throughout the analysis by two-way ANOVA followed by Tukey’s post hoc test).
MeHg exposure induces CHOP expression in the cerebral cortex and striatum of mice, accompanied by an increase in apoptotic cells (Nomura et al., 2022). Therefore, we analyzed the effect of CHOP deletion on neuronal cell death in these regions by TUNEL staining. Consistent with a previous study, MeHg exposure significantly increased TUNEL-positive cells in the primary motor cortex and striatum of wild-type mice. In contrast, CHOP deletion did not affect the increase in TUNEL-positive cells upon MeHg exposure (Fig. 3A, B). In addition, MeHg exposure significantly decreased the number of neurons in the primary motor cortex and striatum of wild-type mice; however, CHOP deletion had no effect, similar to the results of TUNEL staining (Fig. 3C, D). These results suggested that CHOP plays little role in MeHg neurotoxicity in vivo.

Effects of CHOP deletion on methylmercury (MeHg)-induced neuronal cell death. Representative images of (A) TUNEL staining (red) and (C) immunostaining for neuronal nuclei (NeuN) in the primary motor cortex (MO) and striatum (STR). Nuclei were counterstained with DAPI (blue) for TUNEL staining. Wild-type (WT) and CHOP-knockout (KO) mice were administered either 0 ppm (vehicle) or 30 ppm MeHg for 8 weeks. Quantification of (B) TUNEL-positive cells and (D) NeuN-positive cells shown in (A) and (C). The vertical axis shown in (D) represents the number of NeuN-positive cells per 250- × 250-µm2 area. Data are presented as the mean ± S.E. (n = 6-11; *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA followed by uncorrected Fisher's LSD test; ns, not significant). Scale bars represent 50 μm.
The activation of the UPR in response to ER stress ultimately induces a variety of transcription factors to restore cellular homeostasis through the regulation of gene expression. However, the functions of individual transcription factors, especially in the induction of apoptosis triggered by excessive ER stress, remain unclear. Interestingly, MeHg can induce or activate transcription factors other than CHOP, including c-JUN and XBP1 downstream of IRE1α, ATF4 downstream of PERK, and ATF6f (the cytoplasmic domain of ATF6) (Usuki et al., 2008; Usuki et al., 2013; Hiraoka et al., 2017; Hiraoka et al., 2021; Yang et al., 2022). In addition, 4-phenylbutyrate, a chemical chaperone, significantly suppressed apoptosis by inhibiting the upregulation of c-JUN and XBP1 upon MeHg treatment (Pan et al., 2022; Yang et al., 2022). Therefore, ER stress is still considered to be one of the most potent factors for MeHg-induced apoptosis. Based on the results of this study, CHOP-independent transcriptional regulation was expected to contribute to MeHg-induced apoptosis. Further studies focusing on the regulation of gene expression are needed to understand the mechanisms and molecules that induce apoptosis (Fig. 4).
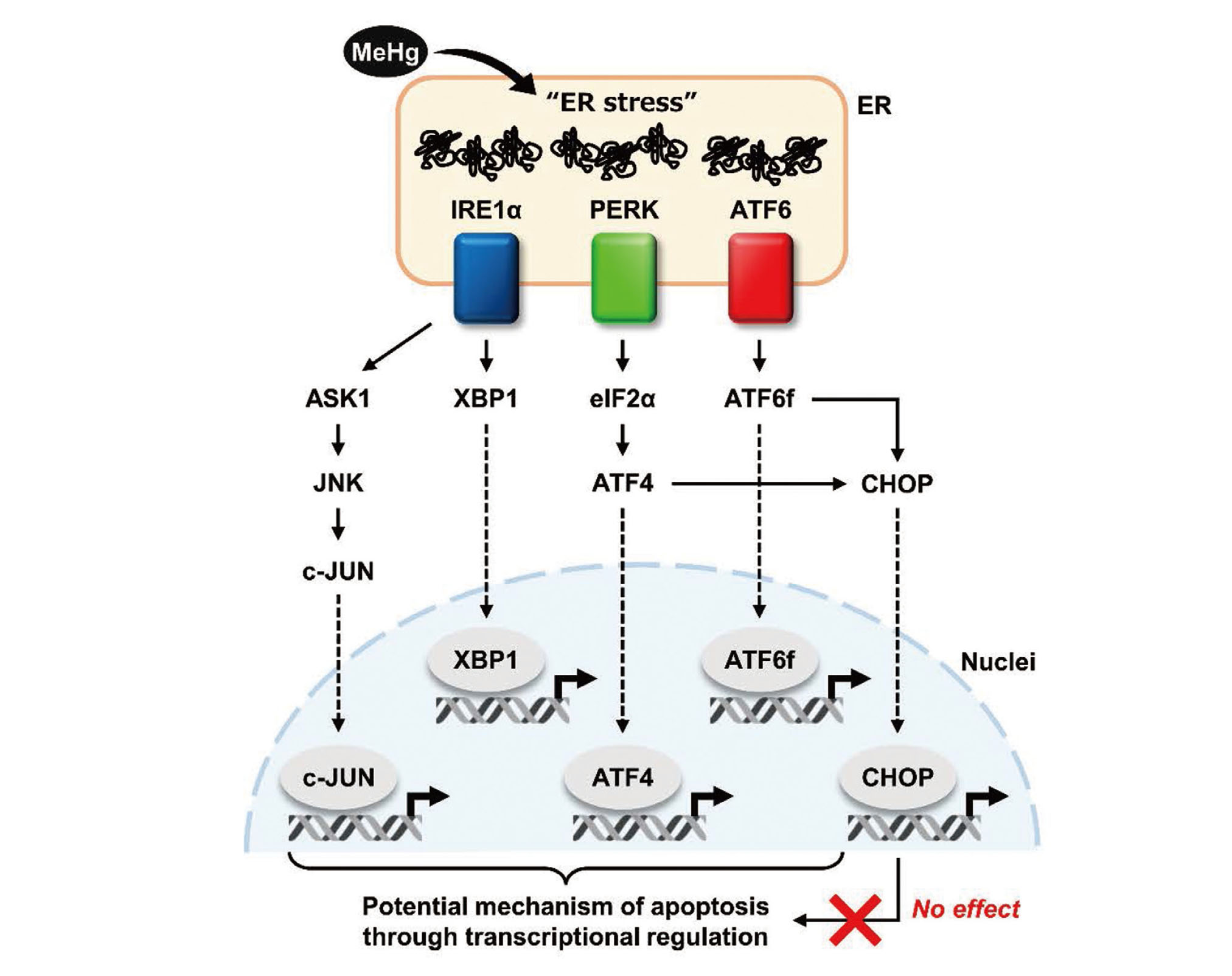
Schematic representation of the UPR-mediated transcriptional regulatory network. This study suggests that the CHOP-dependent pathway is not involved in MeHg neurotoxicity.
Our results suggest that neuronal apoptosis proceeds in MeHg intoxication by a different mechanism than in brain ischemia or 6-OHDA-induced Parkinson's disease models and provide important insights into the mechanism of neuronal cell death triggered by MeHg-induced ER stress.
We thank Kotoe Sueyoshi, Mari Matsumoto, Michiko Fuchigami, Harumi Moriyama, and Runa Shinmura for their excellent technical assistance. This work was supported by JST establishment of university fellowships towards the creation of science technology innovation (JPMJFS2128; to Yuta Iijima) and Study of the Health Effects of Heavy Metals Organized by Ministry of the Environment, Japan (to Takashi Uehara).
Conflict of interestThe authors declare that there is no conflict of interest.