Abstract
A variety of dosage forms have been developed in order to achieve effective and safe drug delivery in topical or systemic drug administrations. In this review, formulation research and process issues related to a popular oral dosage form, the tablet, are introduced. Research on oral dosage forms, including orally disintegrating tablets (ODTs) and films (ODFs), which have recently been developed with an aim toward more patient-centric drug therapy, is also introduced. Another trend in recent drug therapy is an increase in the number of large bioactive molecules among the newly developed active pharmaceutical ingredients (APIs). To design dosage forms for these APIs, novel dosage form design and administration routes are required. For this purpose, we have tried to effectively use the polymer-coated liposomes in oral, pulmonary and ophthalmic administration. For example, suitable polymers were introduced for the design of specific administration routes, such as mucoadhesive liposomes for oral administration. The key point in these researches is the particle design for the component particles of final dosage forms, both in the case of coarse powder particle design for formulating solid dosage forms and in the case of colloidal particle design, such as the design of liposomes for peptide drug delivery.
1. Introduction
One of the most important roles of dosage forms is to deliver the drug into the body. The most common delivery route is that of oral administration, in which the drug is taken by mouth like food and then absorbed in the digestive tract. The active pharmaceutical ingredient (API) is transported to the whole body via the systemic blood stream. Since taking drugs by this route is similar to eating food, the administration can be performed by the patients themselves, and thus oral administration is said to be the method with the best compliance and adherence of the patients. Of course, many other dosage forms are also known, such as injections that are delivered directly into the body, inhalants that allow local administration to the pharynx, and ointments or patches that are expected to act mainly on the skin. These various dosage forms and their names are defined in the pharmacopoeia in each country. In the current (17th edition) Japanese Pharmacopoeia, various dosage form names are listed in each administration routes.
One of the reasons why many dosage forms are required is that the optimal and most efficiently administered dosage form must be designed according to the characteristics and therapeutic purpose of the API. Furthermore, in recent years, the concept of dosage form design for drug administration has evolved, with increasing consideration for the patients who are actually administered the drug—that is, the design of patient-centric dosage forms. Even in oral administration, which is the most preferable administration method for patients, there is an increasing demand for formulation development so that administration can be carried out more easily. For example, orally disintegrating tablets (ODTs) that disintegrate in the oral cavity before swallowing without or with a small amount of water are more convenient for patients. Jelly-like formulations and oral films have also been developed to promote ease of swallowing and administration.
One of the motivations underlying the development of ODTs and other easy-to-take dosage forms is the growing need for medical treatment of the elderly in aging societies, but a growing interest in child-friendly administration modalities has also been a factor. Regarding the former, age-related aspiration is a life-threatening problem, and aspiration pneumonia is ranked as the leading cause of death among the elderly. Although aspiration applies to all situations, including diet, the risk of aspiration should be reduced as much as possible in the administration of therapeutic drugs. In recent years, there has also been a great deal of interest in drug administration to children. When there is no suitable tablet for pediatric dosing, tablets are crushed in the pharmacy to match the prescription. In addition to the labor this requires on the part of the pharmacist, certain functions inherent to the formulation design, such as sustained release, may be impaired by such pulverization. In addition, the division may reduce the accuracy of the drug dose. Even if the pulverization proceeds without such problems, there is a concern that the receiving child may not be able to take the powder well or may refuse to take it entirely due to its unpleasant taste. Against this background, the development of pediatric content formulations as well as the development of dosage forms that facilitate administration to children are underway. Examples of such dosage forms include syrups that have been known for a long time, jelly preparations that are newly listed in the Japanese Pharmacopoeia, and oral film preparations. Also, regarding the size of tablets, a mini tablet with a diameter of about 1 to 4 mm is available as one of the new dosage forms.
The administration of drugs is classified into two types: systemic administration, in which a drug is delivered systemically through the bloodstream after administration, and topical administration, in which a drug is administered directly to the site where its effects are needed. In either case, it is often necessary to deliver the drug into the body more efficiently by using suitable dosage forms and formulations. Sustained-release preparations, in which the drug is gradually released, are one of the delivery modes considered in the preparation design. Orally administered sustained-release preparations control the amount of drug released on the preparation side to control the amount of drug delivered systemically. As a result, specific effects are possible, such as preventing the blood concentration of the drug from reaching a region where side effects may occur, and being able to increase the dose and thus reduce the number of doses. Coating is a typical preparation technique for achieving release control from the dosage forms. A matrix-type tablet, which is prepared with a material suitable for sustained release as the excipient, is also a commonly used dosage form design that achieves release control (McGinity, 1997).
In the case of designing oral preparations of the most common small molecule drugs, it is necessary to pay close attention to the solubility of the drug. In oral administration, the first barrier for delivery into the body is the gastrointestinal tract, and thus the drug must dissolve quickly before absorption there. A drug to be absorbed must pass through epithelial cells in the gastrointestinal tract, and some hydrophobicity of the drug molecules contributes to this permeability. On the other hand, high hydrophobicity of the drug molecules will lead to weak solubility of the crystal. It is well known that this balance between solubility and permeability needs to be taken into consideration (Amidon et al., 1995), and for some time attempts have been made to improve the solubility of poorly soluble drugs in formulation. Such efforts have included miniaturization of drug crystals and solid dispersion technology, and these preparation technologies have been applied to many actual preparations. In the former case, it can be easily imagined that the effective surface area at the time of dissolution is increased and the dissolution characteristics are improved by miniaturization. However, considering the individual processes in the actual manufacture of the final product, mixing the finely pulverized powder into the formulation may lead to problems, including problems with mixing uniformity or a decrease in fluidity during transportation. The latter method, i.e., solid dispersion, has the advantage of potentially achieving greater saturation solubility compared with miniaturization and thus has been anticipated as a useful technology for more than two decades. The biggest issue is the storage stability of the drug in its amorphized state, and the choice of carrier used for the solid dispersion and the mechanism of the amorphization are related.
In formulation design, along with paying attention to the characteristics of the drug crystals, the technology for forming the final dosage form is an important research subject. For example, even in the case of tablet preparation, it is essential to study particle design and formulation design based on the characteristics of the materials (excipients, disintegrants, binders, etc.) that make up the tablet shape having the pharmaceutically required functions. In addition, when special functions such as a significant reduction in disintegration time are required, such as for orally disintegrating tablets, which have been actively developed in recent years, more multifaceted research is required than for the prescription design of ordinary tablets. We have also conducted research on particle design related to tablet excipients, particle design of excipients and binders useful for the formulation design of orally disintegrating tablets, and analysis of compression molding processes using such particles. In developing new dosage forms such as oral film preparations (orodispersible films) and jelly preparations that are expected to have similar functions, we have been working on the basic formulations and the processes of formulation design and preparation methods. For convenience of handling and portability, we have also reported the concept of easily re-dispersible powdered emulsion of oily drugs (dry emulsions) (Takeuchi et al., 1991) or powdered jellies (dry jellies), which can be easily prepared by adding water before use (Kakino et al., 2017).
On the other hand, in order to guarantee the quality of final products, it is necessary to examine the validity of the production process. In addition, it is important to confirm the production process using the actual formulation on a production process scale, because the occurrence of unexpected locking obstacles such as sticking during compaction of pharmaceutical powder formulation poses a major obstacle to production. To address these issues, we have tried to analyze the compression process using a tableting analyzer to detect tablet failures, and to understand the relationship between particle characteristics and sticking phenomena. In addition, these studies have led to the recent study of continuous production. Research on these formulation processes is essential for pharmaceutical companies and has been extensively conducted, but so far, there have been only a small number of papers concerned with conventional formulation research.
Targeting, in which the drug is selectively delivered the desired site in the body, is a field of pharmaceutics research. The development of a drug delivery system that uses submicron-sized carrier particles to deliver drugs to the target site started at late 1970’s (Couvreur et al., 1979). Liposomes were also anticipated to be candidates for particulate drug carriers (Knight, 1981). These fine particles for drug delivery are sometimes called colloidal drug delivery systems (Kreuter, 1994). Active targeting, which utilizes the affinity of substances localized on the cell surface to achieve particle accumulation at the delivery site, has been of interest to many researchers. On the other hand, after administering such submicron particles into the blood by injection, it is necessary to prevent their distribution to other unintended organs or sites for as long as possible. This has been especially important in the development and commercialization of liposome injections containing anticancer agents such as doxorubicin. The same principle applies to the case of topical administration, and the effect of the encapsulated drug depends on how the movement of the microparticulate preparation is controlled after administration. Looking at the development status of drugs in recent years, the size of drug molecules has clearly been increasing. In particular, drugs modeled on hormones, peptides, antibodies, etc., produced in the body have a molecular weight of several thousand to several hundred thousand daltons. Similar to targeting, research on such drugs using a fine particle carrier to deliver them into the body is also in progress.
Insulin is a typical drug with a high molecular weight. Insulin is a substance that is originally produced in the body and has the function of lowering the glucose concentration in the blood when its level is high. Insulin developed as a commercial product has saved the lives of many diabetic patients. For type I diabetic patients who are deficient in insulin, administration before each meal, i.e., just before the glucose concentration in the blood rises, is the most effective treatment method. Injection is the most popular method of delivering insulin into the body, and self-injection is also permitted. In addition, a prefilled syringe-type of insulin in which the injection solution is pre-set in a syringe has also been made available. The pain of injection is alleviated by making the diameter of the needle sufficiently small. A unit called a gauge (G) is used for the thickness of the injection needle: the larger the G value, the thinner the needle. The needle for insulin injection is usually 30 G (outer diameter, 0.31 ± 0.02 mm) or more, which is considerably thinner than the needle used for normal blood sampling (21–23 G: outer diameter, 0.81–0.64 mm). This is another example of pharmaceutical development research progressing in consideration of the needs of actual patients.
Injections are the most common dosage form for high molecular weight drugs, and the development of more patient-friendly optimal dosage forms is still being considered as an important research topic. In the case of insulin, attempts to develop administration methods other than injection have included the commercial development of a powder inhalant to deliver insulin into the body via absorption from the lungs. We have also conducted research focusing on liposome particle design with the aim of developing novel delivery methods other than injection of macromolecular drugs. Among them, polymer-coated liposomes with a mucosal adhesion function have potential for use in the development or oral administration preparations of high molecular drugs. In addition to oral administration, the usefulness of drug transpulmonary absorption using liposomes has also been investigated. We have also suggested the possibility of using eye drops to deliver the drug to the posterior segment of the eye.
As mentioned above, there are various aspects to pharmaceutical formulation research. All the research we have conducted in this field has a common theme: particle design. Such design can range from powder particles to nanoparticles such as liposomes. That is, regardless of the final dosage form, the goal has been to control the characteristics of the basic powder particles and nanoparticles in order to develop the pharmaceutical function and contribute to drug delivery. Moreover, keeping the goal of administration to humans in mind, we have aimed to design dosage forms that will realize the gentlest possible drug administration in actual patients. The author would like to introduce some of the representative researches and their background.
2. Formulation design for improving drug dissolution and absorption
2.1 Micronizing drug crystals and dosage form design
It is well known that the size of drug crystals affects the dissolution rate of drugs (Takeuchi, 2019). As the drug dissolution rate is an important factor in preparing pharmaceutical tablets, it is not rare to pulverize or control the drug crystal to an appropriate size in the manufacture of tablets. In the case of pharmaceutical preparation of poorly water-soluble drugs, a jet mill is often used in pulverizing pharmaceutical crystals. Many active ingredients are organic powders and generally do not have a very high melting point. Therefore, when pulverization to a few microns is required, there is often a concern that the crystal shape may change due to heat generated during pulverization, and thus a jet mill that is relatively unlikely to generate heat is used.
Drug crystals that have been refined to submicron size (usually 100–200 nm in average particle size) are called nanocrystals, and the state in which submicron-sized drug crystals are dispersed in a liquid is called nanocrystal suspension. In addition to improving the dissolution properties, submicron-sized particles exhibit gastrointestinal retention due to tissue adhesion (Ponchel et al., 1997). It has also been reported that the nanocrystal suspension of drug improved its absorption extensibly in comparing AUC (area under curve) values compared with the usual preparation (Liversidge and Cundy, 1995). Regarding the preparation methods, there are roughly two types of methods for refining drug crystals to submicron size: the breakdown method and the build-up method. Well-known breakdown technologies are 1) the pearl milling (bead/ball milling) method (Merisko-Liversidge et al., 2003), which prepares particles by crushing them using a large number of beads in a medium, and 2) the high-pressure homogenization method (Müller and Peters, 1998; Keck and Müller, 2006), which prepares micronized particles by passing them through narrow gaps at high pressure. These breakdown-type processes also have problems such as relatively long operation time, concern about contaminants generated from the medium, and high energy requirement. Elan’s NanoCrystal® technology based on a breakdown method is known a commercially widely used one.
Drug crystals that require improved dissolution properties often have problems with wettability, and even if they are micronized by pulverizing, the effective specific surface area at the time of dissolution does not increase as expected. Conversely, the micronization may cause aggregation and lead to a decrease in the effective surface area for dissolution. To solve this problem, we designed composite particles containing disintegrants by use of a spray drying technique (Takeuchi et al., 1987a). When a poorly water-soluble model drug tolbutamide and PCS® (partially pregelatinized starch), the latter of which has high sphericity, were formulated as disintegrants, the resulting spray-dried particles were spherical and showed excellent fluidity. Because the powder X-ray diffraction analysis of spray-dried particles showed the existence of drug crystals on the particles and the particle sizes of spray-dried particles were comparable to those of PCS particles, the structure of the spray-dried particles was estimated to consist of the drug microcrystals embedded on the surface of PCS, as shown in Fig. 1(A). The dissolution test revealed that the obtained composite particles had significantly improved solubilizing property compared to the original drug crystalline powder and the spray-dried drug alone (Fig. 2). The drug release rate depended on the drug formulating ratio to core particle, PCS. When observed under a microscope, the drug embedded particles were found to swell rapidly upon contact with water, indicating that the prescribed disintegrant worked effectively. A preparation of ordered mixture-like composite particles of poorly-water soluble drug and hydrophilic excipient particles such as Neusilin® (Fig. 1(B)) was also demonstrated with a dry mixing process by using a vibrating ball mill and a mechanical fusion system (Mechanofusion®). The resultant dissolution profiles of the poorly water-soluble drug indomethacin (IMC) from the composite particles were 3-fold to 4-fold faster. It is presumed that the particles having excellent hydrophilicity increased the hydrophilicity of the drug crystals by coating their surface and increased the effective surface area in drug dissolution.


As an attempt at nanocrystal preparation by the build-up method, we tried to control the crystallization of a poorly water-soluble drug using a Pure Nano high-pressure crystallizer (manufactured by Microfluidics) in which the poor solvent addition precipitation method was applied to the precision mixing technique using a high-pressure homogenizer (Takeuchi and Onodera, 2015). By increasing the pressure of the reaction field, a high-strength shear field is formed inside the reaction vessel, and the reactants are mixed in a turbulent state in which the strong energy dissipation mechanism is activated. In the case of crystallization, it was expected that a smaller size of crystals could be obtained by this method compared with the usual mixing operation with a normal stirrer. After preparing a fine crystal suspension of phenytoin as a model of a poorly water-soluble drug and polyvinyl alcohol (PVA) as a dispersion stabilizer, the crystal form, solubility, absorbability after oral administration, etc., of the crystals were evaluated (Tahara et al., 2016). In the case of high-pressure crystallization, the particle size was about half of that in the case of normal crystallization. Moreover, there was no change in the crystal form. The solubilizing property of the prepared particles was remarkably improved. When each phenytoin sample was orally administered to rats and the bioavailability was compared by the drug concentration in blood, the AUC value in the high-pressure crystallization product administration group was 2-fold higher than that of the usual crystallization product.
2.2 Solid dispersion particle prepared by spray drying
The term solid dispersion of a pharmaceutical preparation refers to a preparation in which the drug is dispersed in a solid carrier in an amorphous state. More precisely, the drug is dispersed as a molecule or a cluster in the carrier solids. It is well known that drugs in the amorphous state have excellent dissolution properties (Takeuchi, 2019). However, the amorphous state is generally unstable, and crystallization during storage is one of the strictly required points for pharmaceutical preparations. From this point of view, the selection of the carrier to be blended is important, and many studies have been conducted to determine the best carriers. Furthermore, it has been pointed out that the preparation process in which the drug is first dissolved and the solvent is distilled off, has an energy problem. However, in recent years, the proportion of poorly soluble drugs among the drugs developed has been extremely large, and improvement of solubility has become an extremely important issue. Thus, solid dispersions are one of the important methods for improving the dissolution property of poorly water-soluble drugs.
Research on solid solutions is often said to be the first research on solid dispersion systems (Sekiguchi and Obi, 1961; Chiou and Riegelman, 1969; Simonelli et al., 1969), but research on pharmaceutical preparations has been actively conducted since the 1980s. In order to amorphize drug crystals, it is important to use an appropriate carrier, and water-soluble polymers such as polyvinylpyrrolidone (PVP) have been studied as useful carriers (Corrigan et al., 1983; 1984; Ford et al., 1986). We selected a spray-drying technique to prepare solid dispersion particles, because the resultant particles are spherical and tend to be freely flowing. As a new carrier for preparing the solid dispersion particles prepared by spray-drying, we used fumed silica (Aerosil®), which is insoluble in water but extremely hydrophilic, and is prepared by the spray method (Takeuchi et al., 1987b).
Enteric coating polymer (Eudragit L and hydroxypropyl methylcellulose phthalate (HPMCP)) was also examined as a candidate for carriers. An attempt was made to prepare solid dispersion particles by dissolving dilute ammonia water in which the model drug tolbutamide was dissolved and dispersing or dissolving each carrier. As a result of evaluating the spray-dried particles prepared with a 1:1 ratio of drug: silica by powder X-ray diffraction, it was clarified that the drug exists in an amorphous state in the particles. Crystallization of the drug was observed when the prescribed amount of silica for the drug was reduced, indicating that the interaction between the drug and silica particle surface contributed to the amorphization. All of the particles obtained from the characteristics of the spray drying process were spherical and had excellent fluidity. As shown in Fig. 3, the fastest drug dissolution was confirmed for the solid dispersion particles prepared with a 1:1 ratio of drug: silica, in which complete amorphization of the drug was confirmed. When the ratio of silica was reduced, the elution rate was delayed according to the ratio, and at a ratio of 5:1 it became almost the same as the raw crystalline of tolbutamide.

In order to confirm the usefulness of hydrophilic water-insoluble particles as a carrier of solid dispersion, we also evaluated porous silica (Sylysia®) prepared by a wet method (Takeuchi et al., 2004a). When the ethanol solution of tolbutamide was spray-dried alone, it became a metastable crystalline form II. In the evaporator method, when the solvent was distilled off, it became a stable crystalline form I, suggesting that the resultant crystal form depends on the drying rate. The spray-dried solid dispersion particles of tolbutamide with porous silica were found to be in metastable form II. The metastable crystals with silica in the solid dispersion particles were maintained even during the storage test period, confirming the importance of the presence of the carrier. In addition, the dissolution of tolbutamide in the solid dispersion particles is significantly faster than that of form II particles prepared by spray-drying without silica. The hydrophilicity of the carrier silica improves the wettability of the solid dispersion particles, resulting in the good dissolution.
In the case of indomethacin (IMC), an amorphous state was obtained by spray drying its ethanolic solution regardless of the presence or absence of a carrier. As for the dissolution characteristics, the solid dispersion particles using the porous silica Sylysia350 as a carrier showed extremely rapid dissolution (Fig. 4). When similar indomethacin solid dispersion particles were prepared using porous silica having different pore diameters as a carrier, an amorphous drug was confirmed with any of the silicas. However, when the dissolution rates were compared, it was observed that the drug dissolution was higher in the case of Sylysia®350, which has a larger pore size than Sylysia® 740 (Takeuchi et al., 2003b). When the specific surface areas of the two solid dispersion particles were measured, the value decreased remarkably in the case of Sylysia740, and it was presumed that the pore entrance was closed by the precipitated drug. Therefore, it was concluded that porous silica with a large pore diameter effectively utilizes the pores to amorphize the drug. It is important to pay attention to the prescription ratio of silica and the drug as a carrier and the type of silica, but it has been clarified that porous silica is useful as a carrier of a solid dispersion.

3. Design of orally disintegrating tablets and orally disintegrating films
3.1 Tablet preparation and particle design of excipients
Tablets are usually prepared by compression molding of powder (compaction). Compaction consists of four steps: filling → compression → depressurization → discharge. The powder is discharged from the hopper into the die or into the feeder around the die and then filled in the die. A stirring feeder may be used for discharge from the hopper, but the filling in the die is usually performed by the weight of the powder itself. Powder fluidity is an important factor in these processes. In the next step, i.e., the compression process, the particles are deformed by pressurization to increase the binding force and form a molded product (tablet). Various factors are involved in the binding force between particles. Because the binding force of a substance by van der Waals force is dominant, when the number of bonding points between particles in a tablet is increased, the contact area at the bonding points will be increased, resulting in a stronger molded body. Therefore, particles with large plastic deformability are suitable for compaction. On the other hand, particles with large elastic deformability are not suitable for tablet preparation.
In addition to drug powders, various additive powders (excipients) are used in the preparation of pharmaceutical tablets. One of the most important excipients is filler powder, which has a major influence on compactibility and thus must be designed with care. The commercialization of filler powders for direct tableting began in the 1960s. Lactose is a representative case of a filler that was introduced in the 1960s and has undergone substantial improvement. Lactose is one of the most widely used filler powders for preparing pharmaceutical tablets. The improved compactibility of spray-dried lactose (DCL11) is due to amorphous part of lactose containing 15–20 % in the particle. Anhydrous β-lactose (DCL21), which has better compactibility than a monohydrate (α-lactose) was also commercialized. Over the decades that followed, the development of excipients progressed. As shown in Table 1, many co-processed products of lactose and other substances have been developed as products.
Table 1
Co-processed excipients with lactose.
| Trade Name |
Composition |
Manufacturer |
| Ludipress |
Lactose, PVP*1 (7 %) |
BASF |
| Cellactose |
Lactose, Cellulose (25 %) |
Meggle |
| Pharmatose DCL 40 |
β-Lactose, Lactitol (5 %) |
DMV Veghel |
| MicroceLac |
MCC*2, Lactose |
Meggle |
| StarLac |
Lactose, Maize starch |
Roquette |
*1 PVP : polyvinyl pyrrolidone
*2 MCC: micro crystalline cellulose
The amorphous forms affect the compressibility properties of powders as well as the dissolution property of the resultant dosage forms (Pikal et al., 1978; Vromans et al., 1986). However, the amorphous forms of solids can be easily transformed into more stable, crystalline forms by heat and moisture. Regarding amorphous lactose, studies on its compactibility and the stability of amorphous substances have been conducted from various viewpoints (Sebhatu et al., 1994; 1997; Buckton and Darcy, 1996). Stubberud and Forbes (1998) showed the effects of the physical addition of PVP to amorphous lactose on the crystallization of the amorphous lactose by using a gravimetric system for monitoring the moisture sorption. Saleki-Gerhardt and Zografi (1994) studied that crystallization of amorphous sucrose in the presence of other saccharides, such as lactose, trehalose significantly extended the induction time for crystallization of sucrose.
We evaluated the properties of the particles obtained by prescribing NaAlg (sodium alginate) as a water-soluble polymer in the range of up to 50 % for the purpose of stabilizing amorphous lactose (Takeuchi et al., 1999c; 2000b; 2000c). Fig. 5 shows the results of the thermal stability of amorphous lactose measured by DSC. When no NaAlg or 1 % NaAlg was added, exothermic peaks indicating the crystallization of amorphous lactose were observed. On the other hand, no exothermic peak was detected in the composite particles containing 10 % or more of NaAlg, indicating that the thermal stability was greatly improved. This improvement in thermal stability was found to correlate with an increase in the glass transition temperature (Tg) of lactose detected from the DSC chart. It has been reported that crystallization of amorphous lactose occurs when the Tg value decreases below the storage temperature due to the “plasticizing” effects of water molecules absorbed in lactose (Stubberud et al., 1996). The SD composite particles showed that higher relative humidity values were required for the initiation of crystallization depending on the content of the sodium alginate: 57 % relative humidity for 10 % sodium alginate content and 60 % relative humidity for 30 % sodium alginate content. This increase in the relative humidity required to initiate the crystallization can be attributed to either an increase in the Tg of the composite particles or the absorption of moisture into the sodium alginate.
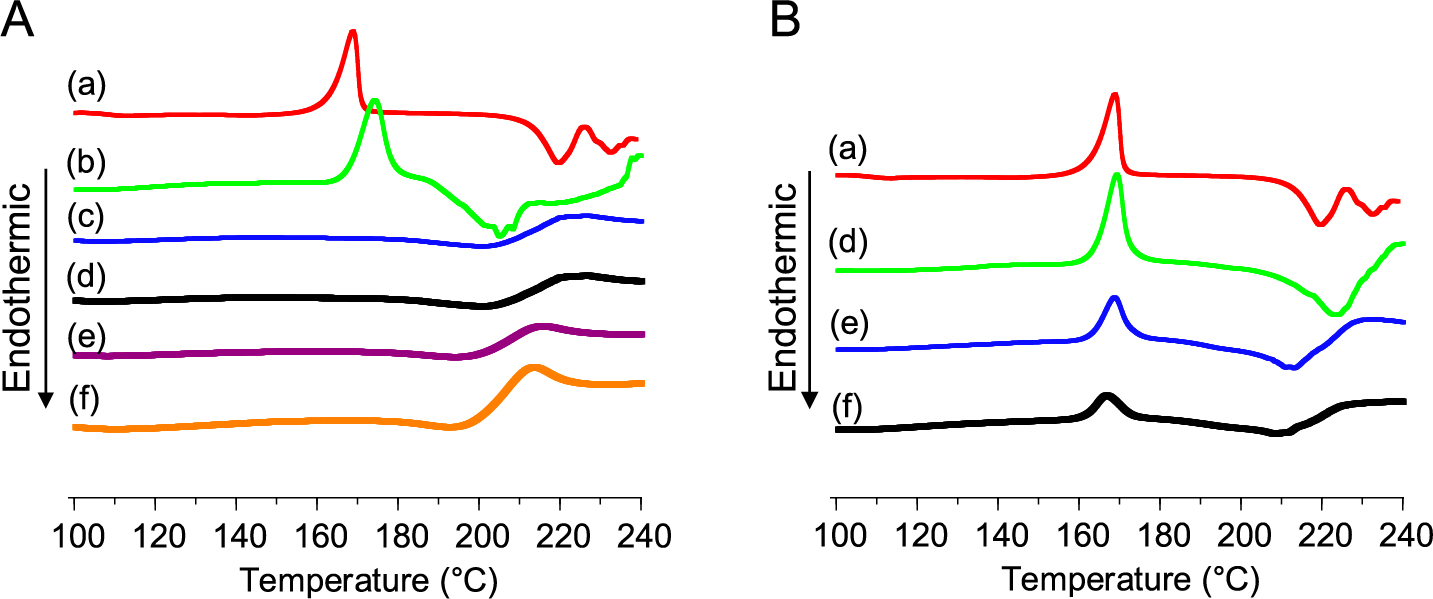
When the compactibility of the composite particles containing amorphous lactose and NaAlg was evaluated, as shown in Fig. 6, excellent compactibility similar to that of amorphous lactose was confirmed up to an amount of 10 % of NaAlg added. However, when the amount of NaAlg was increased to 30 %, a significant decrease in tensile strength was observed, indicating a decrease in compactibility. This change in compression characteristics was well correlated with the change in Tg due to the compositing of NaAlg. The increase in Tg due to NaAlg compositing indicates that the particles become rigid. In fact, when the surface of the tablet is observed with a scanning electron microscope, the fusion of the particles is apparent for the particles with 10 % NaAlg added when compacted under the same conditions, while the shape of the individual particles is almost completely retained in the particles with 30 % addition of NaAlg (Fig. 7). The reason that the Tg of lactose in the particles affects the moldability is that when Lac-NaAlg is stored in a humidified state, the Tg decreases and the hardness of the molded product also increases. This result shows that the excellent plasticity of amorphous lactose decreases depending on the type and amount of the polymer added, and as a result, the hardness of the molded product is greatly reduced. It has been shown that the type and content of the substance to be composited are important factors in order to stabilize the amorphous body while maintaining the high moldability characteristics of the amorphous lactose.

In the design of orally disintegrating tablets (ODTs), various studies have been conducted for the purpose of achieving a tablet that disintegrates extremely rapidly in the oral cavity before swallowing. The first commercialized product, Zydis®, was molded by lyophilizing a solution or suspension of a drug and excipients in a mold (Seager, 1998). Zydis® does not have the strength required for ordinary tablets; rather, the tablet form is maintained by the container. Much research has also been done with the aim of preparing ODTs by using conventional tableting instruments (Sugimoto et al., 2006; Kuno et al., 2008; Okuda et al., 2009). One of the methods is molding a powder layer under wet conditions at a low pressure and then drying it to obtain a tablet hardness close to that of a normal tablet. Another method is to use maltose in the tablet formulation and crystallizing it by humidification and drying treatment after molding to obtain interparticle binding force. A third approach to increase the tablet hardness is to exploit the difference in the melting point of saccharides, and to heat the molded product at an appropriate temperature to promote the crystallization of only one sugar. All three methods have contributed to development of commercial ODTs.
As the demand for ODTs increases, technological development of a method for preparing ODTs using a normal tableting machine is required. Particle design of excipients is important for the preparation of ODTs by conventional tableting processes. Mannitol is most often used as an excipient to obtain rapid disintegration properties. Erythritol is also an excellent excipient candidate because of its solubility and refreshing sensation when dissolved. However, both are inferior in compactibility to lactose, which is most commonly used in ordinary tablets. In the case of erythritol, a molded product cannot be obtained even by compression at extremely high pressure.
For nearly a decade now, we have been conducting research for the purpose of improving the moldability of mannitol and erythritol. As shown in Fig. 8, we determined that constituents of low formable powder, such as mannitol and erythritol, can be spray-dried together with porous silica to form a composite particle, and then an appropriate amount of this composite particle can be mixed with the original low formable powder to dramatically increase the compactibility (Takeuchi et al., 2012). Fig. 9 shows the results of tablet hardness achieved using this composite particle method. It is well known that the hardness of tablets is increased by adding an appropriate amount of silica to the tablet formulation, but in the case of mannitol, such an effect is hardly observed. When our new approach was used to blend composite particles, the tablet hardness became as high as that of typical commercial tablets. For erythritol, which cannot be compression-molded by itself at a high pressure, the tablet hardness could be increased to some extent by mixing silica and separately spray-dried erythritol and tableting. By using the composite particle method, the resultant tablet hardness was much improved. We have also used the composite particle method to examine the formulation of specific ODTs. After adding various types and amounts of disintegrants (2, 5, 10 % w/w) to the basic formulation (a mixture of erythritol, erythritol-silica composite particles (10 % w/w) and magnesium stearate (0.2 % w/w)), a tableting test was performed. All the formulations with the various disintegrants achieved good tablet hardness, but the disintegration time of tablets varied greatly depending on the type of disintegrant. When the disintegrant was cross povidone, the resultant tablet showed the best disintegration property at about 15 seconds.
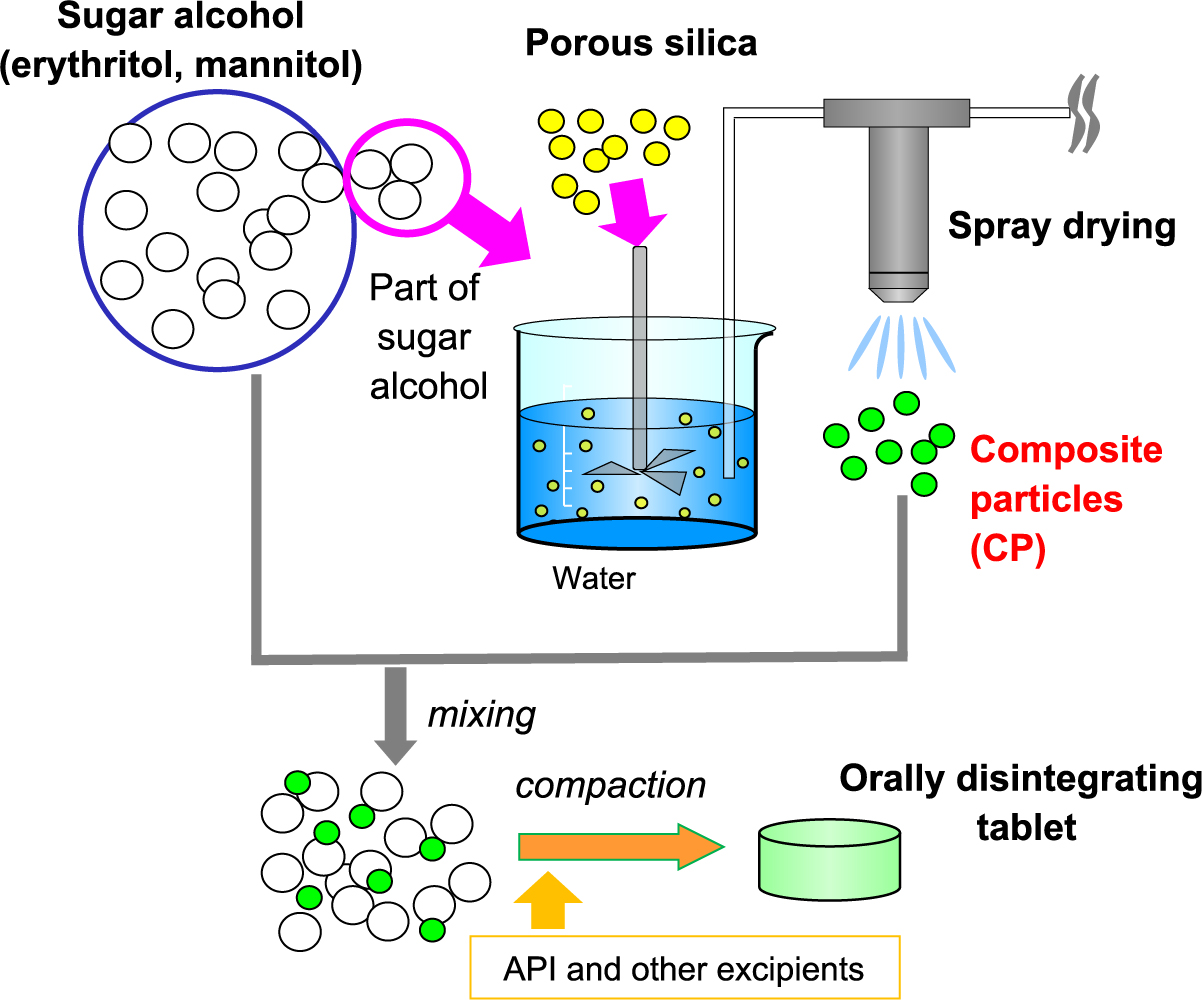
When the crystallinity of erythritol in the composite particles of porous silica-erythritol was evaluated to characterize this composite particle method, the powder X-ray diffraction evaluation indicated no loss of crystallinity, but the endothermic peak due to melting measured by DSC shifted to the low temperature side (Tanimura et al., 2015). This suggested that the crystal energy was high. Moreover, since the proportion of high-energy crystals and the degree of improvement in compactibility were well correlated, it was concluded that this high-energy erythritol contributed to the improvement in compactibility. In fact, when the silica: erythritol ratio was changed to 1:1, 1:2, or 1:3, the 1:2 composite particles showed the highest tablet hardness.
Like lactose, mannitol has also been studied as an excipient, and product development centered on the granulation method has been conducted. So far, several types of mannitol for direct tableting have been made commercially available from several excipient manufacturers. Scanning electron micrographs of the mannitol particles are shown in Fig. 10. In each case, the component is pure mannitol, but the production method alternates between spray-drying and spray-drying granulation. Some particles, such as Mannit Q, have a characteristic particle structure such as a spherical hollow, but the appearance of the other particles in the electron microscopic photographs does not differ substantially (Fig. 10). Crystallinity is almost the same. These commercial products are larger than mannitol crystals and have an improved compactibility. As for Mannit Q, it is presumed that the compactibility at compression is improved because the Mannit Q particle is composed of fine mannitol crystals, as can be confirmed by electron micrographs. Excipients for direct tableting require both high fluidity and excellent compactibility. When the fluidity of some mannitol products for direct tableting was evaluated by the Carr’s index, it was confirmed that all of them had characteristic values suitable for direct tableting.

Incorporation of a binder into a tablet formulation to improve its compactibility is a common formulation design method. In the formulation design of orally disintegrating tablets, it is necessary to pay sufficient attention to the type and prescription amount of the binder for ODTs in order to obtain the tablet characteristic of an extremely short disintegration time. Increasing the amount of binder increases tablet hardness, but leads to a delay in disintegration time. This is considered to be due to water penetrating the tablet, causing the binder to dissolve and adopt a viscous gel state, which delays the penetration of additional water into the tablet. For example, hydroxypropyl cellulose (HPC), which is a pharmaceutical additive widely used in Japan, has several grades used for granulation as a binder. The grade with low viscosity achieved by decreasing the molecular weight is suitable for rapid disintegration of tablets. When using HPC as a binder for the mixing of other powders, the smaller the particle size of HPC, the better the dispersibility in the prescription powder, and thus the greater the number of bonding points between the powders and the greater the hardness of the tablets. We prepared fine particles of HPC using the spray-drying method and the freeze pulverization method. By controlling the particle size of HPC to an average of about 20 μm or less, tablets having the desired characteristics could be prepared. The hardness of the tablets increased remarkably when the spray-dried spherical HPC fine particles were formulated into a tablet formulation (Fig. 11) (Tanimura et al., 2006). In addition, by prescribing the lowest molecular weight and thus lowest viscosity HPC-SSL fine particles as a binder in a model ODT formulation, the desired disintegration of less than 30 seconds and a sufficiently high tablet hardness for practical application could be achieved.

To realize an easier prescription design of ODTs, particle design research has progressed in diverse directions. Development of commercial products such as premixed filler formulations using other additives or composite particles have been produced by excipient manufacturers to allow direct tableting of ODTs using a tableting machine like those used for ordinary tablets. Table 2 lists some of the commercial products and their components.
Table 2
Premixed or co-processed excipients with mannitol for direct tableting of ODT.
| Trade name |
Formulation |
| F-MELT |
Mannitol, xylitol, microcrystalline cellulose, cross povidone, Metasilicate alumic acid magnesium or Anhydrous calcium hydrogen phosphate |
| SmartEx |
Mannitol, L-HPC, PVA |
| Ludiflash |
Mannitol, Collidone CL-SF, Collicoat SR30D |
| Pearitol FLASH |
Mannitol, corn starch |
| Granfiller D |
Mannitol, carmelose, cross-povidone |
L-HPC: Low-substituted hydroxy propyl cellulose
Oral film preparations are preparations having the same characteristics as ODTs. Accordingly, they are sometimes referred to as ODFs (orally disintegrating films). These films are usually stamp-sized and thin as paper. Product development in Europe and the United States has been more active than in Japan, and also the number of products is larger there. In recent years, patient-centric concept has received much attention in dosage form design. Some of pharmaceutical companies are taking account of ODFs as one of the useful dosage forms for sick or pediatric patients who have difficulty in swallowing. In terms of regulation, the ODF is one of the subclasses of dosage forms to be orally administered in the 17th edition of the Japanese Pharmacopeia.
In parallel with the research on ODTs, we have focused on ODFs as a dosage form having the same or more patient-friendly functions. A water-soluble polymer is used as a base corresponding to a filler for tablets; so far, pullulan and hydroxypropyl methylcellulose (HPMC) have been widely used as base polymers for commercial products. In conducting a formulation study of ODFs by using HPMC as a base in the film formulation, we found that the disintegration time could be dramatically shortened by prescribing a small amount of micronized disintegrant low-substituted hydroxypropyl cellulose (L-HPC) as a disintegrant (Takeuchi et al., 2013). It was also shown that the disintegration characteristics could be improved by using insoluble fine particles such as silica and microcrystalline cellulose as well as L-HPC with different particle sizes and shapes in the film. (Takeuchi Y. et al., 2019). The larger particles had a greater impact on disintegration, compared to the smaller particles. However, the addition of larger particles decreased the films’ tensile strength.
For the purpose of searching for an optimum formulation of ODFs, we also investigated the effect of the type of base polymer on the characteristics of resultant ODFs by using hydroxypropyl cellulose (HPC) and polyvinyl alcohol (PVA) as well as the more commonly used HPMC. HPC films were found to have suitable characteristics as a base of ODFs from the standpoints of disintegration and mechanical strength. The HPC films showed particularly good mechanical properties. When model drugs were formulated, these characteristics remained (Takeuchi Y. et al., 2018). HPC is also formulated in some commercially available films prepared with HPMC, probably as a plasticizer in the HPMC film formulation. As the mixing ratio of HPC to HPMC is not disclosed, we examined the mixing ratio of HPC and HPMC. While adding a small amount of HPC (< 10 %) yielded films with a good plasticity, the mechanical properties of the films were significantly deteriorated when increasing amounts of HPC were prescribed to HPMC, such as when a 1:1 ratio of HPC:HPMC was used. This may be attributed to poor compatibility between the polymers. PVA was highly flexible when made into a film and had high refraction resistance. Since it is sticky, it is necessary to take measures to mediate the stickiness. However, it was also confirmed that the film base was suitable for drug encapsulation, because its mechanical property values were not easily altered, even if a drug is added in the formulation. (Takeuchi Y. et al., 2020a).
4. Progress on the solid formulation manufacturing process
4.1 Analysis of the powder compaction process
The powder properties required for the formulation of tablets with the desired functions were described above in the sections 3.1 and 3.2. However, the tableting process also has a great influence on the preparation of the compacted product, and thus various studies have been conducted to examine these effects of tableting. There are two main approaches for the preparation of tablets: (1) direct tableting, in which the drug powder and other excipient powder are mixed, and (2) indirect tableting or granule compression, in which part or all of the powder is granulated and compressed in advance. In either case, the compaction process consists of filling the die with powder, compressing, and then discharging the tablet from the die after releasing pressure. A stress-displacement curve (Fig. 12(A)) can be drawn by precisely measuring the displacement during compression and the upper punch load over time for obtaining information on the compression characteristics of the powder. Depending on the type of powder, the displacement is reduced (expansion of the compacted product) after the displacement is maximized at the maximum pressure of the upper punch. This is due to elastic recovery of the compacted powder. The net energy required for compaction is calculated by subtracting the energy for elastic recovery (B) from the total energy required for molding (A + B). The net energy is expressed as a ratio to the total energy and used as an index of compactibility. The compression equation most often used when evaluating compression molding characteristics using a compression model is the Heckel equation (1) (Heckel., 1961a; 1961b).

Here, ɛ is the porosity of the powder layer, P is the compression pressure, and the pressure from the upper punch is usually used for this measurement.
As shown in Fig. 12(B), when the compression pressure is increased, causing the compression to proceed and the powder layer to begin undergoing plastic deformation, the increase on the left side of the Heckel equation becomes constant and appears as a straight line. This slope is k, and the reciprocal of the slope is used as the yield pressure (Py) as an index of plastic deformation.
The compaction property of powders has been evaluated with various parameters such as yield pressure measured by Heckel analysis (Humbert-Droz et al., 1983), stress relaxation (van der Voort Maarschalk et al., 1997) and elastic recovery (Picker, 2001). In general, a powder with plastic deformability is preferable for preparing a compacted product as it increases the tablet hardness, while the property of elasticity works in the opposite way.
It is possible to estimate one of the tablet failures in tableting, capping, by using these parameters. Capping, in which the contact surface of the tablet is peeled off with the punch, is thought to be caused by any of several different aspects of the compaction process. Higher elasticity of compacted powder is one of the main factors for capping during compaction. The measurement of die wall pressure may also be a useful method to evaluate the compaction property of powders, because residual die wall pressure reflects the plastic property of the compacted powder in the die (Carless and Leigh, 1974). Some papers have reported that the residual die wall pressure is one of the main factors for capping, with other factors being the elastic recovery during decompression, the stress concentration due to die wall pressure at decompression (Hiestand et al., 1977) and the entrapped air in the tablet during the compaction process (Tanino et al., 1995). Sugimori reported that capping occurs when a tablet is cracked by residual die wall pressure at the final stage of the decompression process (Sugimori et al., 1989a; 1989b).
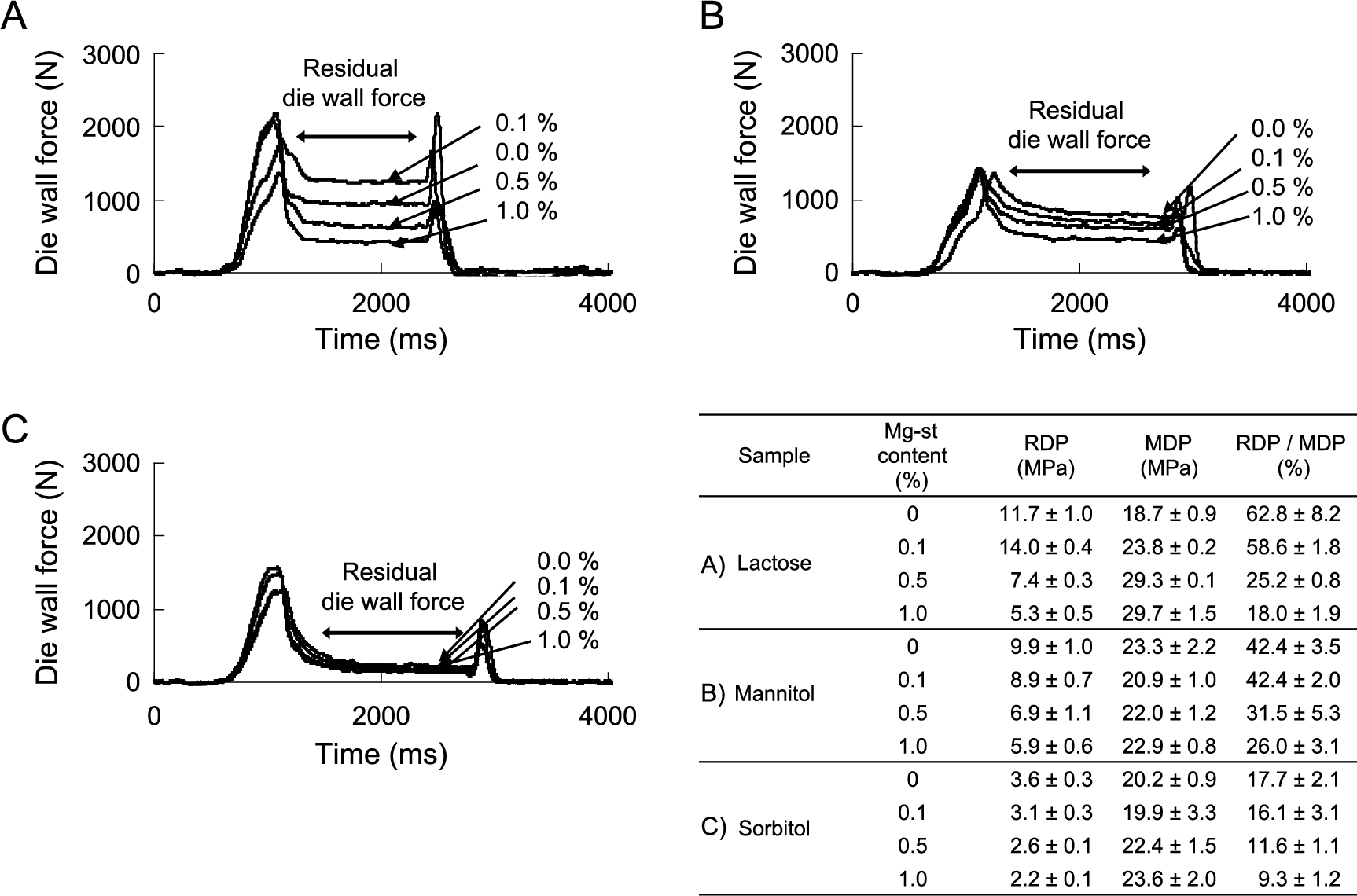
In the studies mentioned above, the compression parameters were measured by using static compression with a very slow compression speed. We evaluated the compression molding process using a compaction analyzer (Tab All®) that can measure the pressure detected at each site on a single-shot tableting machine for analysis in a dynamic compressed state similar to an actual tableting machine. Fig. 13 shows an example of the data under various loads during compaction. The load applied to the wall surface increases with the increase of the upper punch load and shows the maximum die wall pressure (MDP), and then the pressure is reduced and the force detected even after the pressure is completely removed is called the residual die wall surface pressure (RDP). The MDP value increased when the compression pressure was changed with the same powder. In addition, since the MDP value differs greatly depending on the powder even at the same compression pressure, it was proposed to evaluate by the RDP/MDP value standardized by the MDP rather than directly comparing the RDP value. The RDP/MDP value reflected the compacting property of pharmaceutical powders. For example, crystalline lactose and ascorbic acid, which showed a capping tendency, showed relatively larger RDP/MDP values (Takeuchi et al., 2004b). Fig. 14 shows the results of measuring the residual die wall pressure profile by changing the amount of the lubricant for lactose, mannitol, and sorbitol, which are excipients of three typical pharmaceutical tablets (Takeuchi et al., 2005b). Sorbitol, which has the best compactibility, showed a low residual wall pressure regardless of the prescription amount of the lubricant. Mannitol, as well as lactose, tends to cause tablet failures such as binding and capping depending on the tableting formulation and conditions. For example, if the prescription amount of the lubricant in the tablet formulation increases, the tablet failure tendency is depressed. The decrease in RDP/MDP value with increasing the amount of the lubricant was well correlated to this tendency.
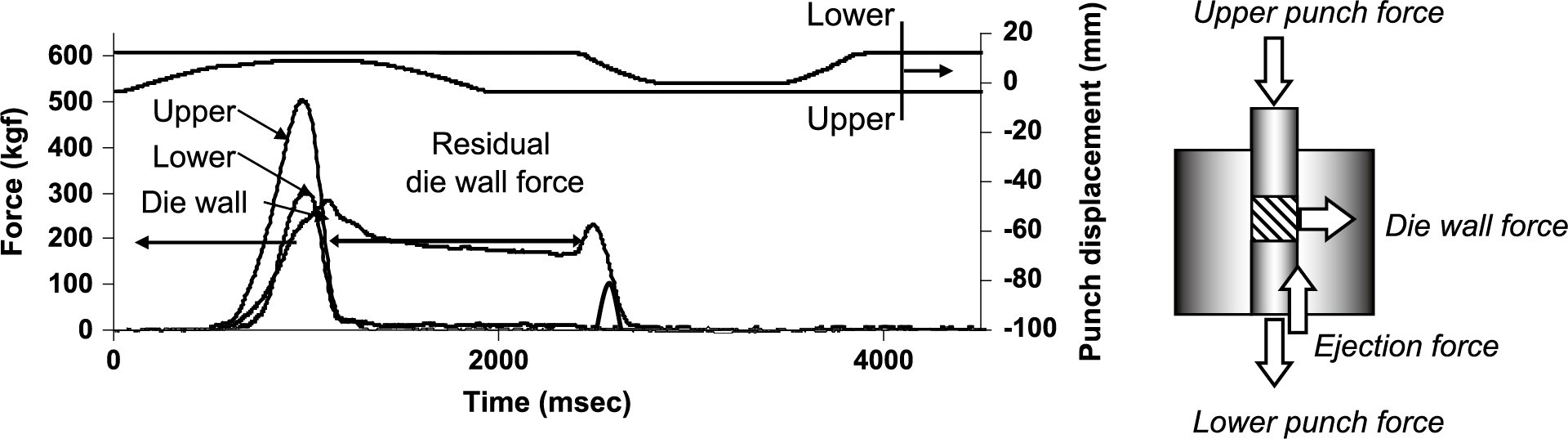
In recent years, for the purpose of more practical analysis of tableting processes, a tableting simulator (Style One®) capable of compression molding in a form closer to that of a rotary tableting machine was developed. Not only does the rotary tableting machine used for production have a faster compression molding speed than the single-punch tableting machine, but also the size of instrument, the installation angle of machine parts such as rotating discs, and often the compression rollers for compression molding (depending on the model of machine) have different characteristics between the rotary and single-punch tableting machine. These differences cause the compression time and the interval of each compression molding process to differ depending on the model of the tableting machine (Narang et al., 2010). In addition, in the process of drug development, it is inevitable that the scale of the tableting machine will change, and it is necessary to set new tableting conditions each time. At that time, process parameters are usually determined based on experience and trial and error, and thus the development of a convenient method for setting the parameters in these cases is strongly required. The tableting simulator developed in recent years can reproduce the tableting process of various rotary tableting machines, and can obtain the compaction parameter values of various rotary tableting machines even though it is a single-punch tableting machine.
Using this tableting simulator, evaluation of the effect of the compression rate on tablet characteristics (Michaut et al., 2010) or evaluation of the pressure applied to the tablet when the tablet is ejected from the die in the tablet ejection process (Sun, 2015) has been reported. Takahashi et al. conducted a series of experiments with the goal of efficiently optimizing the tableting conditions associated with changes in the tableting machine that occur during scale-up and technology transfer. Even if compaction is performed under the same conditions by different types of tableting machines, the tablet characteristics will change. It was found using a tableting simulator that the change in tableting speed was most critical to the change in compactibility and thus tablet properties. As a new prediction model for determining the optimum tableting conditions when changing the tableting machine, it was found that the area under the curve (AUC) in the time-pressure profile considering the compression time is an important parameter (Takahashi et al., 2019). Based on this concept, a model for predicting tableting conditions (locking pressure, rotation speed) was constructed, and its suitability was confirmed by comparing data from different locking machines (Takahashi et al., 2020). Utilizing these parameters, it will be possible to compare parameters in the tableting process to standardize them during scale-up or technology transfer.
4.2 Tableting failures on a production scale
In addition to the capping discussed in section 4.1, several other types of tablet failures occur, such as sticking, binding, and chipping. Among them, sticking, in which a part of the powder adheres to the surface of the punch, may lead to serious problems in the production scale. It is conceivable that the number of tablets will increase, the surface of the punch will deteriorate due to long-term use, and the adhesion of powder will proceed more easily. These sticking obstacles not only affect the product quality, but also lead to damage to the punch due to the application of extremely high pressure to the punch. It seems reasonable to consider that a possible reason for the sticking is that the bonding force between the particles in the powder bed is weaker than that of powder on the surface of the punch. In order to clarify the detailed mechanism responsible for the sticking to avoid this tablet failure, several studies have been conducted. These works have reported various potential causes of the sticking, such as the physical and mechanical properties of active powders (Naito and Nakamichi, 1969; Danjo et al., 1993), water content, hygroscopicity of the formulations, lubricant types and concentrations, (Danjo et al., 1997; Waimer et al., 1999a; Roberts et al., 2004), tableting tools and punch surface shapes, and operating conditions (Newton et al., 2000; Sixsmith and McCluskey, 1981; Waimer et al., 1999b; Kakimi et al., 2010) such as engraving of the punched surface and finishing of the punched surface.
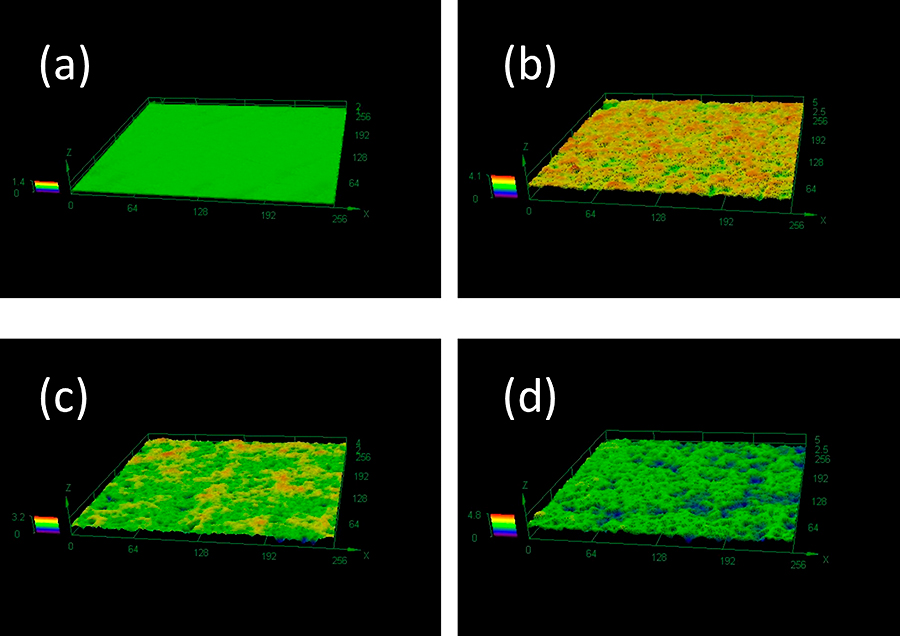
In order to prevent sticking during the production of tablets, it is conceivable that the amount of the binder could be increased or both the amount of the binder and the amount of the lubricant in the tablet formulation could be increased. However, it may be difficult to change the formulation after fixing it for approval. On the production side, attempts will be made to try to control tableting conditions such as the tableting speed or pressure. Recently, tableting machines companies or manufacturers of parts of machines such as punches and dies have proposed alternative methods to avoid these tablet failures. In one case, the surface treatment of the punch was developed for the purpose of suppressing the adhesion between the powder and the surface of the punch and avoiding sticking, and several kinds of punches with different surface treatments are commercially available in Japan. As can be seen from the laser micrograph (Fig. 15), the surface of the punch is uneven, unlike the surface of a normal punch. The concept is that the unevenness reduces the contact area with the powder and reduces adhesion of the powder. We evaluated the effect of a punch surface treatment using these punches and the effect of reducing adhesion using two types of pharmaceutical crystals with different plastic deformability (Takeuchi Y. et al., 2020b). Using a single-shot tableting machine, tableting was performed up to several times, and the amount of drug adhering to the surface of the punch was quantified by HPLC. As a result, in the case of a drug such as acetaminophen, which was evaluated to have low plastic deformability as confirmed by the Heckel plot, the unevenness of the punch surface tended to reduce the drug adhesion to the punch surface (Fig. 16). On the other hand, in the case of ibuprofen, which has a relatively large plastic deformability, the amount of adhered drug was almost the same regardless of the presence or absence of unevenness on the surface of the punch. Rather, the tendency of increasing adhesion of P3, which has the largest unevenness, was observed to be the largest. These results suggested that when the plastic deformability is large, the area of contact of the powder on the uneven punch surface during tableting is large, and thus the adhesion tendency is increased.

As described above, pharmaceutical companies want to avoid problems such as tableting failures during production, and thus predicting such problems at the time of tablet prescription design is an important goal. For this purpose, Pitt et al. (Pitt et al., 2015) conducted a study using the GTP-1® and proposed a criteria for feasibility of tableting with a formulation. The proposed value of the strength of tablet (TFS) as measured by GTP-1 is greater than 2 MPa in tableting at 200 MPa. They also proposed that the discharge pressure (ES) should be smaller than 5 MPa. Osamura et al. (2016) systematically compared the GTP-1 measurement results with the actual tableting results obtained with a rotary tableting machine to determine TFS and ES. They tried to visually express the tableting characteristics of the prescription by classifying them into four areas as an index of compactibility and an index of manufacturability. When the validity of this evaluation system was verified using pharmaceutical excipients with known tableting characteristics, the results reflected the characteristics of individual excipients (Fig. 17). Furthermore, it was also shown that even with a practical formulation, a series of formulations that may have an effect on manufacturability, such as a change in the blending amount of a lubricant, can be examined and the effect can be visually confirmed (Osamura et al., 2017). They selected drugs for which a sticking phenomenon was easily observed, changed the blending amount of lubricant and tablet shape, and compared the results obtained using GTP-1 and a rotary tableting machine. A good correlation between the two was obtained even for tablet formulations in which different amount of the lubricant was formulated and/or for tablets prepared by using different punches having various shapes. Using the same method, it was also demonstrated that this estimation for tableting process using the GTP-1® is feasible for indirect tableting with granulated drugs with excipients to avoid sticking (Osamura et al., 2018).

5. Liposomal particle design for drug delivery
5.1 Polymer-coated liposomes for efficient drug delivery
Phospholipids are constituents of cell membranes. The surface of liposomal particles composed of phospholipid bilayers can be easily modified with both positive and negative charges by prescribing a small amount of charged substance together with phospholipids in the preparation. By prescribing an appropriate amount of cholesterol, the fluidity of and the degree of hydrophobicity of liposomal particles can be controlled by controlling the prescribed amount. Furthermore, it has been reported that the surface of liposome particles can be modified with polymers, surfactants, etc. (Sunamoto et al., 1984). Due to these characteristics, liposomes have been studied as drug carriers for targeting drugs in the body after injection. As described in the introduction of this manuscript, liposomal injections of anticancer agents have been developed as commercial products. Liposomes have also been studied as drug carriers to avoid a barrier for entry into the body. Research aimed at gastrointestinal absorption of insulin is a typical example of the latter type of application, and has been studied by multiple research groups since the mid-1970s (Patel and Ryman, 1976; Dapergolas and Gregoriadis, 1976). However, the usefulness of liposomal formulations for the enteral absorption of insulin has not been confirmed (Patel and Ryman, 1981; Shenfield and Hill, 1982).
We have been conducting studies with a goal of imparting various pharmaceutical functions by controlling the surface properties of liposome particles. Fig. 18 shows two typical polymer-coated liposomes designed for this purpose. One is a comb-shaped coating formed by penetrating a hydrophobic moiety chemically bonded to a water-soluble polymer into a liposome lipid bilayer (the anchor ring method), and the other is a coating created by the electrical interaction between a charged liposome surface and a polymer having the opposite charge. Regarding the former, surface modification was demonstrated using a polymer, PVA-R, in which an alkyl group was introduced at the end of polyvinyl alcohol (PVA). The formation of the thick polymer layer was confirmed by measuring the zeta potential of the liposomes treated with several polymers under different polymer concentrations (Fig. 19). The zeta potential of the negatively charged liposomes became neutral when the polymer concentration was increased. Compared with the liposomes that physically absorbed PVA, the stabilizing effect of the PVA-R-coated liposomes was excellent due to their three-dimensional coating layer (Takeuchi et al., 1994; 1998).
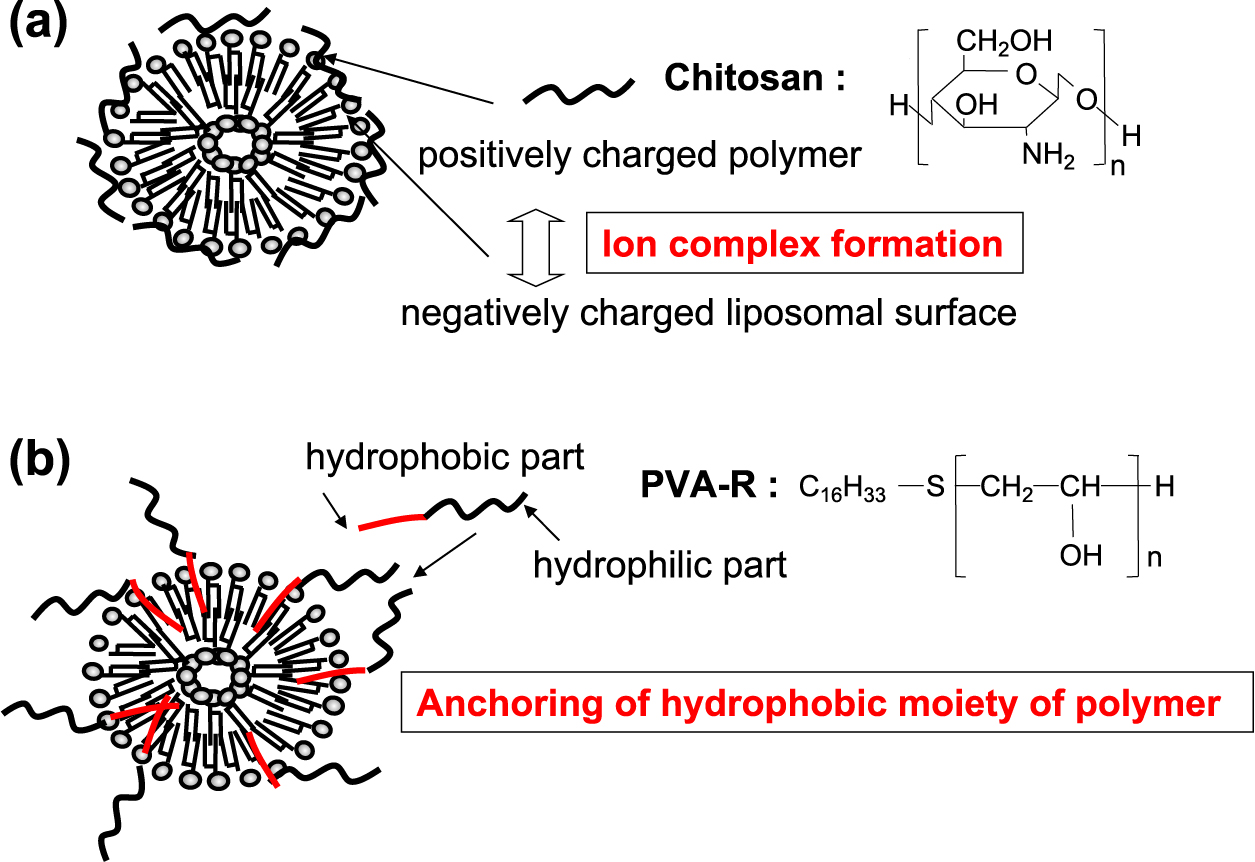
PEGylated liposomes prepared by formulating polyethylene glycol conjugated phospholipids have been studied extensively as targeting carriers for the anticancer drug doxorubicin and commercial products for clinical use are now available (Senior, 1987; Allen et al., 1989; 1991; Papahadjopoulos et al., 1991). PVA-R-coated liposomes were found to have a similar function to PEGylated liposomes, such as improving blood retention after intravenous injection to deliver a drug effectively to cancer tissues in rats (Takeuchi et al., 1999a; 1999b; 2000a; 2001a,b,c). PEG liposomes form a polymer layer on the surface prepared by formulating PEG conjugated phospholipids as a liposomal component, whereas the polymer layer of PVA-R-coated liposomes is able to be formed after preparation of drug loaded liposomes.
5.2 Mucoadhesive liposomes for oral administration of peptide drugs
In order to improve the efficiency of gastrointestinal absorption, it is conceivable to prolong the residence time of the drug or the carrier containing the drug after oral administration. This concept was discussed for potential application to conventional dosage forms such as tablets or granules in the 1980s (Guputa et al., 1990). In order to use liposomes as a carrier for oral insulin administration, we considered improving the retention of liposomal particles in the intestinal tract by allowing their interaction with the mucin layer on the inner surface of the digestive tract.
Polymer-coated liposome particles having a mucoadhesive function were prepared using chitosan (CS), which is prepared by deacetylating chitin and are reported to interact with mucin when used as a topical dosage form (Nagai, 1985). The mucoadhesive property of polymer-coated liposomes was evaluated by using intestinal tubes separated from rats. In this test, the number of liposomal particles withdrawn from the liposomal suspension in the tube was calculated by using a Coulter counter before and after incubation. The extent of adhesion to the mucous layer in the intestinal tube was expressed by the following equation.
|
Adhesive
(
%
)
=
N
0
-
N
15
N
0
×
100 | (3) |
were, N0 is the initial number of liposomal particles and N15 is the number of liposomal particles 15 min after incubation.
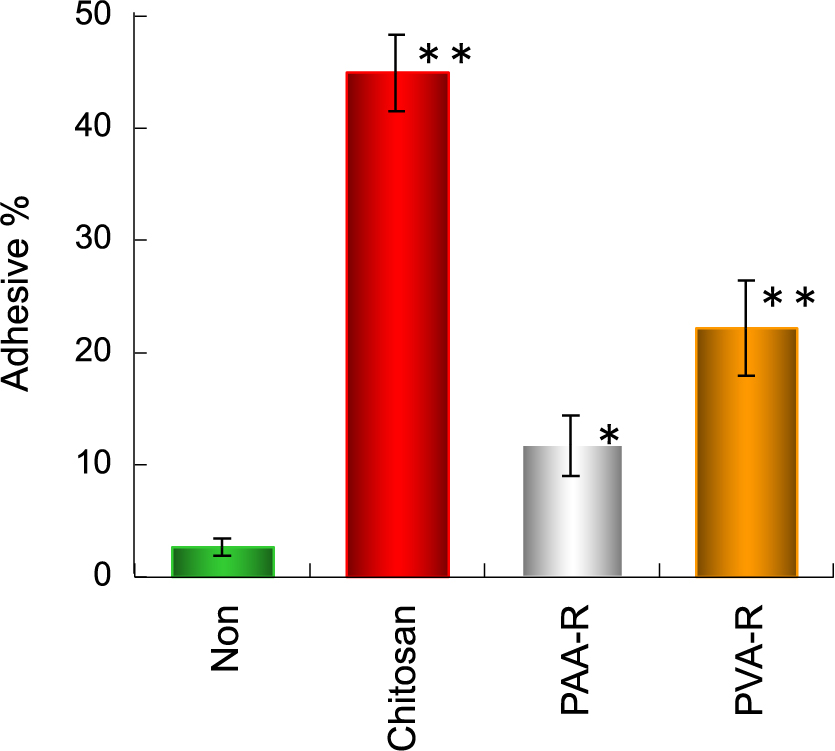
As shown in Fig. 20, the highest adhesion property of CS-coated liposome (CS-Lip) was confirmed in this in vitro test (Takeuchi et al., 1994). The affinity of CS-Lip to the mucosa is presumed to be due to the electrostatic interaction between the positive charge of the amino group of CS and the negative charge of mucin, which is a mucosal component. The physical interaction due to the entanglement of both polymers is also responsible for this adhesion. The latter interaction was supported by the fact that negatively charged acrylic acid polymer (PAA)-coated liposomes also showed the mucosal adhesion properties. It is believed that the acid polymer (PAA)-coated liposomes also showed the mucosal adhesion properties. It is believed that the negative charge of PAA extends linearly without shrinking the polymer and works effectively for entanglement with mucin.
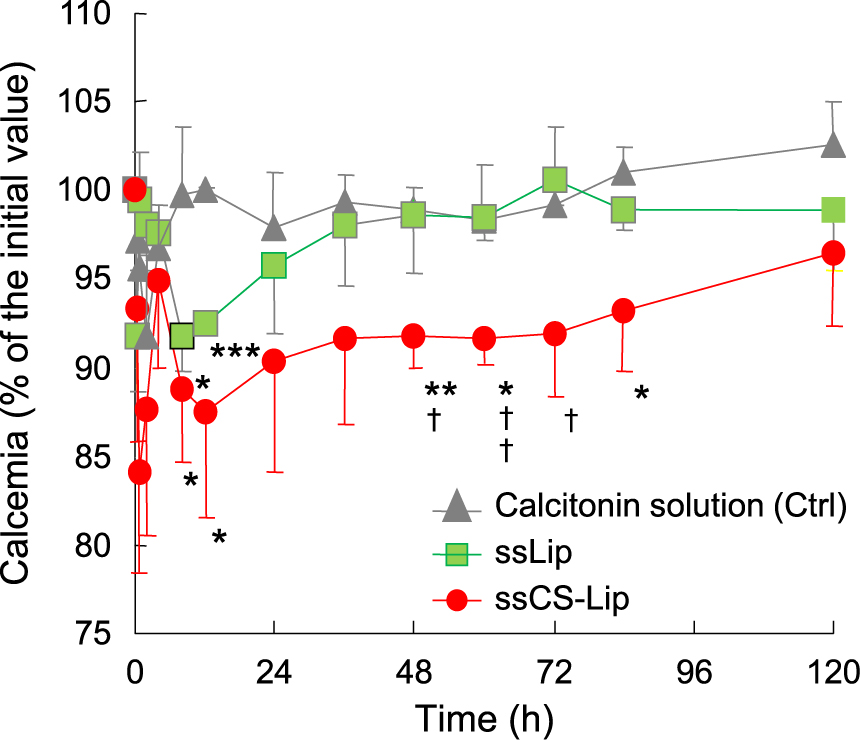
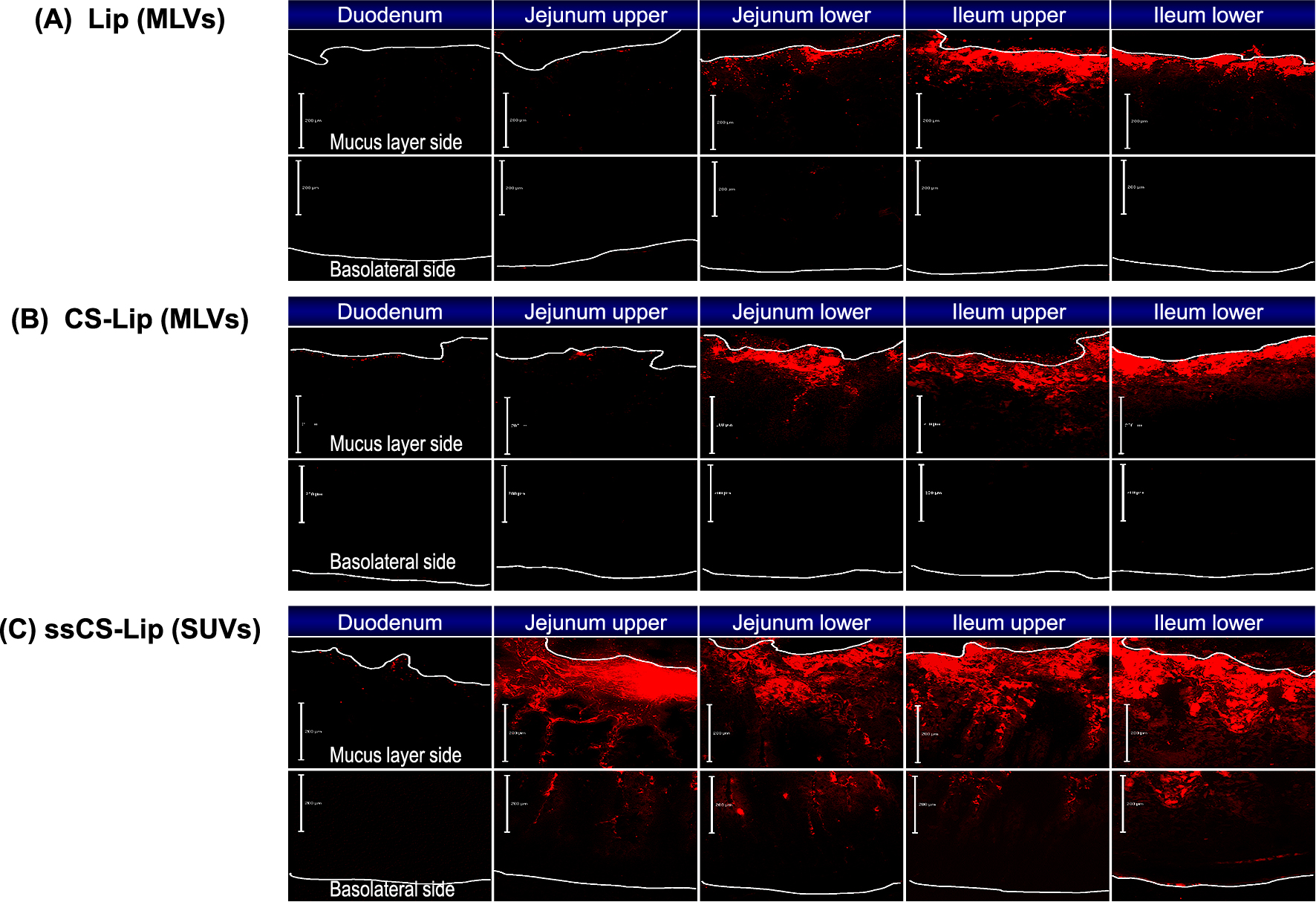
CS-Lip encapsulating insulin was orally administered in rats, and the blood glucose levels were measured to evaluate their effectiveness in enteral absorption. A significant decrease in the blood glucose level was observed after administration of CS-Lip, confirming the enteral absorption of insulin. The significant decrease in value was maintained up to 12 hours, suggesting the retention of the liposomal carrier in the intestinal tract due to the mucosal adhesion property (Fig. 21) (Takeuchi et al., 1996). The importance of the CS layer for mucosal adhesion and resultant insulin absorption was revealed by the fact that no significant effect was observed under the same conditions when liposomes having a positive charge were used as a carrier. The usefulness of polymer-coated liposomes for the oral administration of peptide drugs was confirmed by using another model peptide drug, calcitonin. A single administration of calcitonin encapsulated in CS-Lip lead to a decrease in the blood calcium concentration in rats for a long period of time (Fig. 22) (Takeuchi et al., 2003a). It was also clarified that the particle size is an important factor for drug absorption (pharmacological effect) with mucoadhesive liposomes, because the absorption increased remarkably when the particle size was decreased to submicron size. Since these differences were considered to be related to the behavior of liposomes in the gastrointestinal tract, the intestinal tube was taken out after administration of several types of liposomes containing a lipophilic fluorescent marker in rats and its cross section was observed with a confocal laser scanning microscope. It was confirmed that the CS-Lip had a longer residence time in the intestinal tract compared with the uncoated liposomes, and they adhered more to the upper part of the intestinal tract (Fig. 23(A), (B)). By reducing the particle size of CS-Lip to the submicron size, the number of liposomes retained in the intestinal tube was considerably increased, and the liposomal particles were observed to be penetrated into the mucosa over time (Fig. 23(C)) (Takeuchi et al., 2001d; 2005a). It was also confirmed that CS in the CS-Lip moved in the digestive tract together with liposomal particles (Thongborisute et al., 2006).

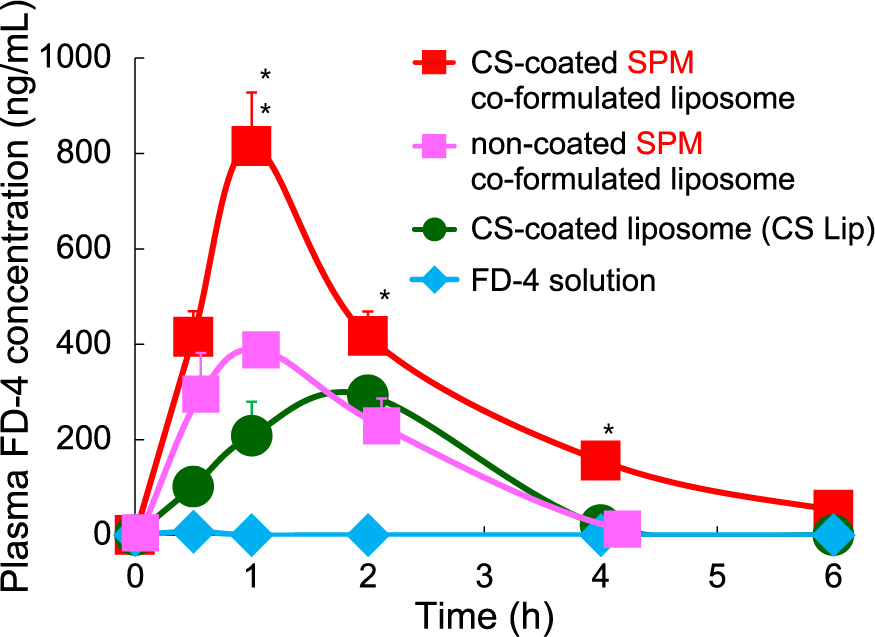
Following the demonstration of the usefulness of liposomes with mucosal adhesion properties for the oral administration of peptide drugs, studies on the characterization of the mucoadhesive properties of polymers were carried out to understand the mucoadhesive mechanism. Novel methods for evaluating mucoadhesive properties of polymers in vitro were also developed. The methods include a simple mucin particles method and Biacoa method for quantitative evaluation (Takeuchi et al., 2005c). In the latter method, an interaction analyzer, Biacoa, based on surface plasmon resonance is used to try to evaluate the mucoadhesive property. We also tried to improve the mucosal adhesive property of polymers themselves by partly modifying the molecules or conjugation of different types of polymers. One of examples is chitosan-aprotinin (Werle et al., 2009a; 2009b). Carbopol is a commercial product of polyacrylic acid, whose mucoadhesive property has been reported. The mucoadhesive property of Carbopol was found to be improved by conjugating with lectin or WGA (wheat germ agglutinin) (Werle et al., 2010; Makhlof et al., 2011a). It was also confirmed that simultaneous encapsulation of water-soluble absorption enhancers into liposomal carriers greatly improved the absorption of calcitonin (Makhlof et al., 2011b). With respect to the absorption control with these liposomal carriers, the mucoadhesive functions of CS-Lips were demonstrated to render them effective as carriers for a small molecule, indomethacin, by measuring the actual absorption amount (Sugihara et al., 2012). Recently, we also confirmed the delivery of a model hydrophilic macromolecule, dextran labeled with fluorescein isothiocyanate (FITC), with an average molecular weight of ca. 4000 daltons (FD-4) or more, to systemic blood flow by absorption in the digestive tract after oral administration with liposomal formulations. The absorption amount was greatly increased by very small when a FD-4 solution was administered. The amount of absorption was greatly increased by simultaneously encapsulating an absorption enhancer, spermine (Fig. 24). The simultaneous administration of spermine dissolved in a FD-4 solution had almost no effect on the absorption, indicating that it is important for the absorption enhancer to be delivered to the absorption site in the gastrointestinal tract together with the absorbed macromolecular drug. These absorption data are well consistent with the pharmacological data shown above in this section.
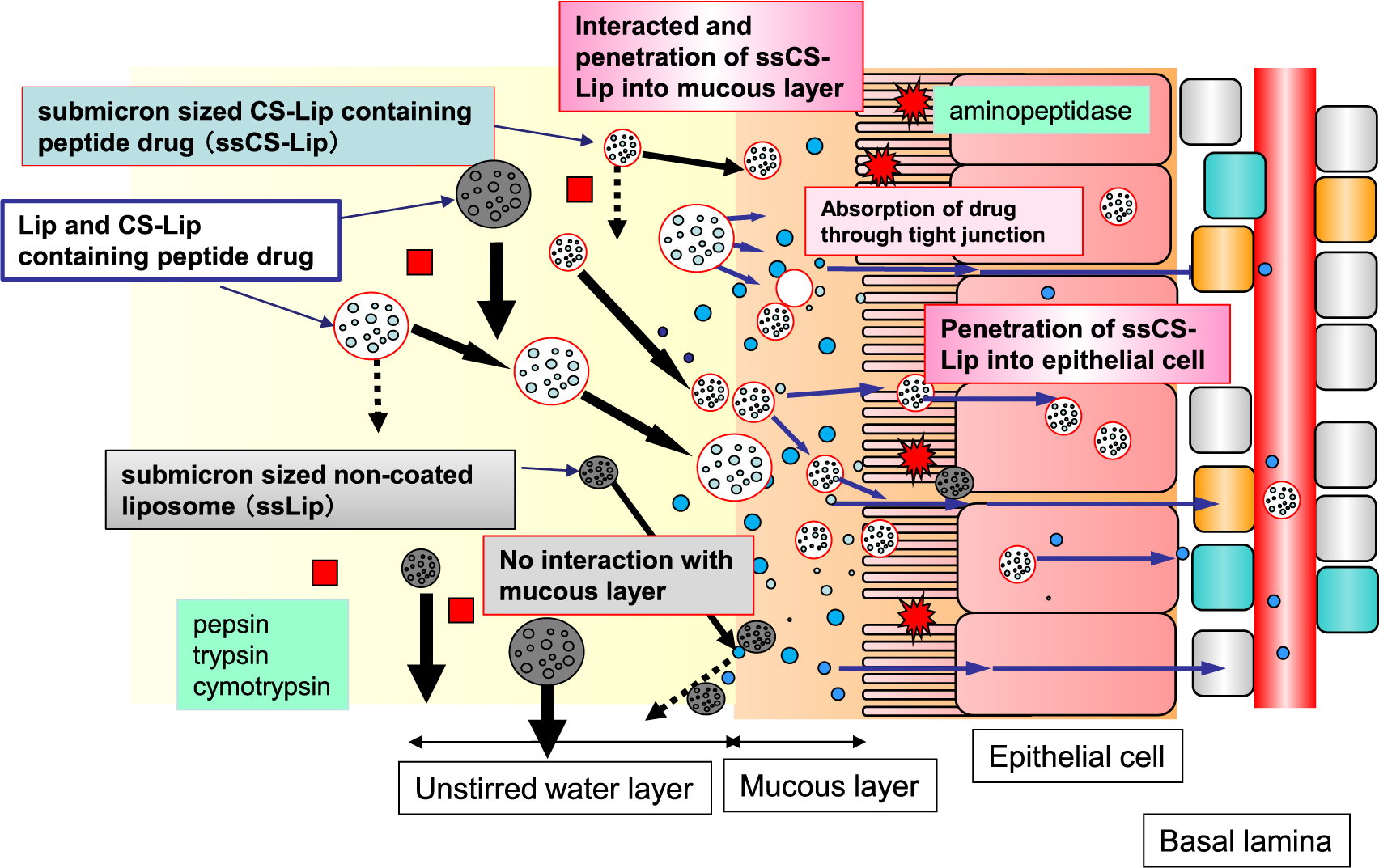
Comprehensively considering the behavior of liposome particles in the gastrointestinal tract and absorption experiments, the absorption of peptide drugs in the gastrointestinal tract by muco-adhesive micronized liposomes represented by submicron-sized CS-Lip (ssCS-Lip) can be represented schematically as shown in Fig. 25. As predicted at the beginning of the study, the mucous membrane (mucosa) presents a large barrier to the absorption of macromolecular drugs in the gastrointestinal tract. CS-Lips interact with and retain the mucous layer on the epithelial cell in the flow of the intestinal tract and penetrate more deeply if the particles are small enough. We speculate that some of the particles are taken up by mucosal epithelial cells as they are. Considering that the absorption enhancer that was administered simultaneously with the particles worked effectively, it can be inferred that some macromolecular drugs are released in the mucous layer and permeate tight junctions from there.
Inhalation systems can be a useful drug delivery method for both topical and systemic administration of drugs. Several types of research have been conducted on such administration systems, because particle design including nanoparticle engineering and characterization is important to achieve the most efficient drug delivery (Johnson et al., 2020; Price et al., 2019). Kawashima et al. have also tried to prepare suitable drug crystals and their formulations for dry powder inhalation systems (Kawashima et al., 1998a; 1998b). In terms of peptide drug administration, the lung is highly expected to be a suitable site for systemic administration of drugs. In fact, Exubera® and Afrezza® have been developed and marketed as powdered inhaled insulin preparations. On the other hand, the lungs can be affected by intractable diseases such as lung cancer and chronic obstructive pulmonary disease, and research on new drug treatments such as nucleic acid drugs is progressing. In such cases, it is highly expected that the development of a suitable delivery system such as liposomes will be utilized as a topical dosage form.
In comparing pulmonary administration with oral administration, the lungs are a closed system and there are few degrading enzymes, so it is expected that pulmonary administration of peptide drugs will lead to absorption. When calcitonin solution and various types of polymer-coated liposomes encapsulating calcitonin were administered intrapulmonarily, the effect of the coating polymer on drug absorption was found to be different from that in oral administration, as shown in Fig. 26 (Murata et al., 2012). There was a difference in the degree, with the highest absorption-enhancing effect being observed for the PVA-R-coated liposomes. We conjectured that the PVA-R-coated liposomes showed the highest absorption because the PVA coating on the liposome avoided phagocytosis by alveolar macrophages and increased the retention time (Nakano et al., 2008). This idea was proved by measuring the retention of these polymer-coated liposomes. In fact, when we evaluated the retention of CS-Lips and PVA-R-coated liposomes in the lungs, we found that a large number of CS-Lips remained in the lung tissue and a large number of PVA-R-coated liposomes remained in the bronchoalveolar lavage fluid (BALF). The finding that the retention of PVA-R-coated liposomes was extended after pulmonary administration was further confirmed by a non-invasive method using an in vivo imaging system and a fluorescence marker, indocyanine green (Murata et al., 2014). In the case of PVA-R, the drug encapsulated in liposomes remained for a longer period of time and then was gradually released into the lungs, which lead to a sustained efficacy. In the case of transpulmonary administration, a trial using the model polymer drug FITC-Dextran confirmed that the absorption actually occurs at the higher level than by oral administration. We have recently confirmed that FITC-Dextran (FD-4), which has an average molecular weight of 4000, was absorbed even when administered as an aqueous solution.

As a clearer example of the role of drug delivery systems in the local administration of drugs, we attempted to deliver a drug to the posterior segment of the eye by an instillation centered on the retina. In recent years, with the rapid aging of the population in Japan, the incidence of posterior ocular diseases such as glaucoma, diabetic retinopathy, age-related macular degeneration (AMD), and retinitis pigmentosa has rapidly increased, such that new and more effective drug therapies are needed. VGF, a growth factor, is known to have an important role in cell generation. An anti-VEGF (vascular endothelial growth factor) that suppresses the generation of new, edema-induced blood vessels appearing near the macula of the retina with high probability has been developed. However, its delivery is generally limited to invasive intravitreal injections. However, eye drops containing the anti-VGEF antibody and exerting suitable anti-inflammatory and antioxidant effects are also available, and if these drops could be reliably delivered to the posterior segment of the eye, they could potentially be used to treat the above-named diseases.
For the purpose of evaluating the transfer of liposome particles into the posterior segment of the eye after instillation, a liposome suspension containing coumarin-6 as a fluorescent marker was instilled in mice, and the retina was observed with fluorescent microscopy. In the case of submicron-sized liposomal particles, the observation of the coumarin-6 color was used to confirm that the particles had reached their target (Fig. 27) (Hironaka et al., 2009). To confirm that the size of the particles affected their ability to move to the posterior part of the eye, a similar evaluation was performed by using liposomal particles having different particle sizes ranging from the micron order to 100 nm. The results showed that the smaller the particle size, the better the delivery efficiency (Inokuchi et al., 2010). Since not only particle size but also the lipid composition was found to affect the ability to reach the target, the hardness of the liposome particles was also one of the factors.

To confirm the clinical effects of liposomal carriers in eye drop instillation, we tried to conduct animal experiments as a joint research project with a laboratory equipped for ophthalmic evaluation. When liposomes encapsulating edaravone, a free radical scavenger, were instilled into a model mouse before light irradiation to induce retinal photodamage, the liposomal formulation was confirmed to be effective at preventing the subsequent photodamage (Hironaka et al., 2011). It was also clarified that diclofenac, which is one of the NSAIDs (nonsteroidal anti-inflammatory drugs), can suppress choroidal neovascularization due to retinal photodamage in mice when instilled as a drug encapsulated in liposomes (Fujisawa et al., 2012; Shimazawa et al., 2017). In either case, such a pharmaceutical effect in the retinal area was not observed when the solutions dissolving these drugs were instilled under the same conditions for comparison, and it could be judged that the drug could be delivered to the vicinity of the retina by the liposomal eye drop preparation. A newly developed macromolecular API was also applied to this liposomal system and confirmed to achieve a higher pharmacological effect in mice (Taketani et al., 2016).
As with oral and transpulmonary administration, an increase in the amount or duration in drug delivering by polymer-coated liposomes was also investigated. Poly-L-lysine (PLL) and polyarginine (PLA), which have been reported to have affinity for cells, were tested as coating polymers, and the delivery efficiency of these polymer-coated liposomes to the posterior segment of the eye was assessed (Sasaki et al., 2013). The results showed that the larger the molecular weight of the polymer, the greater the effect. However, the efficiency decreased owing to the aggregation of liposomes if the molecular weight of the polymer was too large. Thus, an optimum molecular weight existed, and this weight was determined to be 15,000 daltons in the case of PLL. Active targeting with polymer-coated liposomes has been tested in this ophthalmic liposomal system. One of the targets is folic acid receptors, and an enhancement in delivering efficiency has been observed by modifying the surface of liposomes with folic acid. The delivery of macromolecules to the posterior part of the eye has also been attempted by using FITC-dextran as a drug model, and the feasibility of this approach was confirmed. Based on this finding, it is expected that macromolecular APIs such as siRNA and Bonac nucleic acid can also be delivered to the posterior segment of the eye. Further research is ongoing.
6. Conclusion
Research on formulation design and dosage form design for drug delivery were described in this review. Regarding the design of solid dosage forms, the particle design of drug crystals and several excipients have important roles for the objective functions of the resultant dosage forms. It is important to image the objective functions expected by patients as a final goal of research. Regarding the delivery of macromolecular drugs, colloidal drug carriers have important roles, and the concept of particle design is also important even for these colloidal particles. The suitable design of submicron-sized particles such as polymer-coated liposomes not only improves the efficiency in drug delivery as a drug-targeting tool but also leads to the development of new drug delivery methods such as the oral administration of peptide drugs. As briefly discussed in this paper, it is necessary for pharmaceutical researchers to account for the actual needs of patients as well as the needs of the researchers and medical personnel. It is expected that several types of particle design research for a variety of pharmaceutical materials will contribute to the design of truly patient-friendly dosage forms to solve various issues related to the unmet needs of patients.
References
- Allen T.M., Hansen C., Martin F., Redemann C., Yau-Young A., Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo, Biochimica et Biophysica Acta (BBA)—Biomembranes, 1066 (1991) 29–36. DOI: 10.1016/0005-2736(91)90246-5
- Allen T.M., Hansen C., Rutledge J., Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues, Biochimica et Biophysica Acta (BBA)—Biomembranes, 981 (1989) 27–35. DOI: 10.1016/0005-2736(89)90078-3
- Amidon G.L., Lennernäs H., Shah V.P., Crison J.R., A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability, Pharmaceutical Research, 12 (1995) 413–420. DOI: 10.1023/A:1016212804288
- Buckton G., Darcy P., Water mobility in amorphous lactose below and close to the glass transition temperature, International Journal of Pharmaceutics, 136 (1996) 141–146. DOI: 10.1016/0378-5173(96)04503-6
- Carless J.E., Leigh S., Compression characteristics of powders: radial die wall pressure transmission and density changes, Journal of Pharmacy and Pharmacology, 26 (1974) 289–297. DOI: 10.1111/j.2042-7158.1974.tb09278.x
- Chiou W.L., Riegelman S., Preparation and dissolution characteristics of several fast-release solid dispersions of griseofulvin, Journal of Pharmaceutical Sciences, 58 (1969) 1505–1510. DOI: 10.1002/jps.2600581218
- Corrigan O.I., Holohan E.M., Sabra K., Amorphous forms of thiazide diuretics prepared by spray-drying, International Journal of Pharmaceutics, 18 (1984) 195–200. DOI: 10.1016/0378-5173(84)90119-4
- Corrigan O.I., Sabra K., Holohan E.M., Physicochemical properties of spray dried drugs: phenobarbitone and hydroflumethiazide, Drug Development and Industrial Pharmacy, 9 (1983) 1–20. DOI: 10.3109/03639048309048541
- Couvreur P., Kante B., Roland M., Guiot P., Bauduin P., Speiser P., Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: preparation, morphological and sorptive properties, Journal of Pharmacy and Pharmacology, 31 (1979) 331–332. DOI: 10.1111/j.2042-7158.1979.tb13510.x
- Danjo K., Kamiya K., Otsuka A., Effect of temperature on the sticking of low melting point materials, Chemical & Pharmaceutical Bulletin, 41 (1993) 1423–1427. DOI: 10.1248/cpb.41.1423
- Danjo K., Kojima S., Chen C.Y., Sunada H., Otsuka A., Effect of water content on sticking during compression, Chemical & Pharmaceutical Bulletin, 45 (1997) 706–709. DOI: 10.1248/cpb.45.706
- Dapergolas G., Gregoriadis G., Hypoglycæmic effect of liposome-entrapped insulin administered intragastrically into rats, The Lancet, 308 (1976) 824–827. DOI: 10.1016/S0140-6736(76)91209-5
- Ford J.L., The current status of solid dispersions, Pharmaceutica Acta Helvetiae, 61 (1986) 69–88. https://pubmed.ncbi.nlm.nih.gov/3520598/
- Fujisawa T., Miyai H., Hironaka K., Tsukamoto T., Tahara K., Tozuka Y., Ito M., Takeuchi H., Liposomal diclofenac eye drop formulations targeting the retina: formulation stability improvement using surface modification of liposomes, International Journal of Pharmaceutics, 436 (2012) 564–567. DOI: 10.1016/j.ijpharm.2012.07.024
- Gupta P.K., Leung S.-H.S., Robinson J.R., Bioadhesives/mucoadhesives in drug delivery to the gastrointestinal tract, in: Vincent Lenaerts, Gurny Robert (Eds.) Bioadhesive Drug Delivery Systems, CRC Press, Florida, 1990, pp. 65–92, ISBN 9780849353673.
- Heckel R.W., Density-pressure relationships in powder compaction, Transactions of the Metallurgical Society of AIME, 221 (1961a) 671–675.
- Heckel R.W., An analysis of powder compaction phenomena, Transactions of the Metallurgical Society of AIME, 221 (1961b) 1001–1008.
- Hiestand E.N., Wells J.E., Peot C.B., Ochs J.F., Physical processes of tableting, Journal of Pharmaceutical Sciences, 66 (1977) 510–519. DOI: 10.1002/jps.2600660413
- Hironaka K., Inokuchi Y., Fujisawa T., Shimazaki H., Akane M., Tozuka Y., Tsuruma K., Shimazawa M., Hara H., Takeuchi H., Edaravone-loaded liposomes for retinal protection against oxidative stress-induced retinal damage, European Journal of Pharmaceutics and Biopharmaceutics, 79 (2011) 119–125. DOI: 10.1016/j.ejpb.2011.01.019
- Hironaka K., Inokuchi Y., Tozuka Y., Shimazawa M., Hara H., Takeuchi H., Design and evaluation of a liposomal delivery system targeting the posterior segment of the eye, Journal of Controlled Release, 136 (2009) 247–253. DOI: 10.1016/j.jconrel.2009.02.020
- Humbert-Droz P., Gurny R., Mordier D., Doelker E., Densification behaviour of drugs presenting availability problems, International Journal of Pharmaceutical Technology and Product Manufacture, 4 (1983) 29–35.
- Inokuchi Y., Hironaka K., Fujisawa T., Tozuka Y., Tsuruma K., Shimazawa M., Takeuchi H., Hara H., Physicochemical properties affecting retinal drug/coumarin-6 delivery from nanocarrier systems via eyedrop administration, Investigative Ophthalmology & Visual Science, 51 (2010) 3162–3170. DOI: 10.1167/iovs.09-4697
- Johnson L.M., Mecham J.B., Quinn F., Hickey A.J., Nanoparticle technology for respiratory tract mucosal vaccine delivery, KONA Powder and Particle Journal, 37 (2020) 97–113. DOI: 10.14356/kona.2020013
- Kakimi K., Niwa T., Danjo K., Influence of compression pressure and velocity on tablet sticking, Chemical and Pharmaceutical Bulletin, 58 (2010) 1565–1568. DOI: 10.1248/cpb.58.1565
- Kakino Y., Hishikawa Y., Onodera R., Tahara K., Takeuchi H., Gelation factors of pectin for development of a powder form of gel, dry jelly, as a novel dosage form, Chemical and Pharmaceutical Bulletin, 65 (2017) 1035–1044. DOI: 10.1248/cpb.c17-00447
- Kawashima Y., Serigano T., Hino T., Yamamoto H., Takeuchi H., A new powder design method to improve inhalation efficiency of pranlukast hydrate dry powder aerosols by surface modification with hydroxypropylmethylcellulose phthalate nanospheres, Pharmaceutical Research, 15 (1998a) 1748–1752. DOI: 10.1023/A:1011916930655
- Kawashima Y., Serigano T., Hino T., Yamamoto H., Takeuchi H., Design of inhalation dry powder of pranlukast hydrate to improve dispersibility by the surface modification with light anhydrous silicic acid (AEROSIL 200), International Journal of Pharmaceutics, 173 (1998b) 243–251. DOI: 10.1016/S0378-5173(98)00243-9
- Keck C.M., Müller R.H., Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation, European Journal of Pharmaceutics and Biopharmaceutics, 62 (2006) 3–16. DOI: 10.1016/j.ejpb.2005.05.009
- Knight C.G. (Ed.), Liposomes: From Physical Structure to Therapeutic Applications, Elsevier, Amsterdam (1981), ISBN: 9780444803207.
- Kreuter K. (Ed.), Colloidal Drug Delivery Systems., Marcell Dekker, New York (1994). ISBN: 9780824792145.
- Kuno Y., Kojima M., Ando S., Nakagami H., Effect of preparation method on properties of orally disintegrating tablets made by phase transition, International Journal of Pharmaceutics, 355 (2008) 87–92. DOI: 10.1016/j.ijpharm.2007.11.046
- Liversidge G.G., Cundy K.C., Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs, International Journal of Pharmaceutics, 125 (1995) 91–97. DOI: 10.1016/0378-5173(95)00122-Y
- Makhlof A., Fujimoto S., Tozuka Y., Takeuchi H., In vitro and in vivo evaluation of WGA–carbopol modified liposomes as carriers for oral peptide delivery, European Journal of Pharmaceutics and Biopharmaceutics, 77 (2011a) 216–224. DOI: 10.1016/j.ejpb.2010.12.008
- Makhlof A., Werle M., Tozuka Y., Takeuchi H., A mucoadhesive nanoparticulate system for the simultaneous delivery of macromolecules and permeation enhancers to the intestinal mucosa, Journal of Controlled Release, 149 (2011b) 81–88. DOI: 10.1016/j.jconrel.2010.02.001
- McGinity J.W. (Ed.), Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms, 2nd ed., Marcel Dekker, New York (1997), ISBN: 9780824797737.
- Merisko-Liversidge E., Liversidge G.G., Cooper E.R., Nanosizing: a formulation approach for poorly-water-soluble compounds, European Journal of Pharmaceutical Sciences, 18 (2003) 113–120. DOI: 10.1016/S0928-0987(02)00251-8
- Michaut F., Busignies V., Fouquereau C., Huet de Barochez B., Leclerc B., Tchoreloff P., Evaluation of a rotary tablet press simulator as a tool for the characterization of compaction properties of pharmaceutical products, Journal of Pharmaceutical Sciences, 99 (2010) 2874–2885. DOI: 10.1002/jps.22032
- Müller R.H., Peters K., Nanosuspensions for the formulation of poorly soluble drugs: i. preparation by a size-reduction technique, International Journal of Pharmaceutics, 160 (1998) 229–237. DOI: 10.1016/S0378-5173(97)00311-6
- Murata M., Nakano K., Tahara K., Tozuka Y., Takeuchi H., Pulmonary delivery of elcatonin using surface-modified liposomes to improve systemic absorption: polyvinyl alcohol with a hydrophobic anchor and chitosan oligosaccharide as effective surface modifiers, European Journal of Pharmaceutics and Biopharmaceutics, 80 (2012) 340–346. DOI: 10.1016/j.ejpb.2011.10.011
- Murata M., Tahara K., Takeuchi H., Real-time in vivo imaging of surface-modified liposomes to evaluate their behavior after pulmonary administration, European Journal of Pharmaceutics and Biopharmaceutics, 86 (2014) 115–119. DOI: 10.1016/j.ejpb.2013.09.006
- Nagai T., Adhesive topical drug delivery system, Journal of Controlled Release, 2 (1985) 121–134. DOI: 10.1016/0168-3659(85)90038-0
- Naito S., Nakamichi K., Studies on techniques of manufacturing pharmacy. i. prediction of tableting troubles such as capping and sticking. (i), Chemical & Pharmaceutical Bulletin, 17 (1969) 2507–2514. DOI: 10.1248/cpb.17.2507
- Nakano K., Tozuka Y., Takeuchi H., Effect of surface properties of liposomes coated with a modified polyvinyl alcohol (PVA-R) on the interaction with macrophage cells, International Journal of Pharmaceutics, 354 (2008) 174–179. DOI: 10.1016/j.ijpharm.2007.08.046
- Narang A.S., Rao V.M., Guo H., Lu J., Desai D.S., Effect of force feeder on tablet strength during compression, International Journal of Pharmaceutics, 401 (2010) 7–15. DOI: 10.1016/j.ijpharm.2010.08.027
- Newton J.M., Haririan I., Podczeck F., The influence of punch curvature on the mechanical properties of compacted powders, Powder Technology, 107 (2000) 79–83. DOI: 10.1016/S0032-5910(99)00173-4
- Okuda Y., Irisawa Y., Okimoto K., Osawa T., Yamashita S., A new formulation for orally disintegrating tablets using a suspension spray-coating method, International Journal of Pharmaceutics, 382 (2009) 80–87. DOI: 10.1016/j.ijpharm.2009.08.010
- Osamura T., Takeuchi Y., Onodera R., Kitamura M., Takahashi Y., Tahara K., Takeuchi H., Characterization of tableting properties measured with a multi-functional compaction instrument for several pharmaceutical excipients and actual tablet formulations, International Journal of Pharmaceutics, 510 (2016) 195–202. DOI: 10.1016/j.ijpharm.2016.05.024
- Osamura T., Takeuchi Y., Onodera R., Kitamura M., Takahashi Y., Tahara K., Takeuchi H., Prediction of effects of punch shapes on tableting failure by using a multi-functional single-punch tablet press, Asian Journal of Pharmaceutical Sciences, 12 (2017) 412–417. DOI: 10.1016/j.ajps.2017.05.001
- Osamura T., Takeuchi Y., Onodera R., Kitamura M., Takahashi Y., Tahara K., Takeuchi H., Formulation design of granules prepared by wet granulation method using a multi-functional single-punch tablet press to avoid tableting failures, Asian Journal of Pharmaceutical Sciences, 13 (2018) 113–119. DOI: 10.1016/j.ajps.2017.08.002
- Papahadjopoulos D., Allen T.M., Gabizon A., Mayhew E., Matthay K., Huang S.K., Lee K.D., Woodle M.C., Lasic D.D., Redemann C., Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy, Proceedings of the National Academy of Sciences, 88 (1991) 11460–11464. DOI: 10.1073/pnas.88.24.11460
- Patel H.M., Ryman B.E., Oral administration of insulin by encapsulation within liposomes, FEBS Letters, 62 (1976) 60–63. DOI: 10.1016/0014-5793(76)80016-6
- Patel H.M., Ryman B.E., Systemic and oral administration of liposomes, In: C.G. Knight (Ed.), Liposomes: From Physical Structure to Therapeutic Applications, Elsevier, Amsterdam, 1981, pp. 409–441. ISBN: 9780444802347.
- Picker K.M., Time dependence of elastic recovery for characterization of tableting materials, Pharmaceutical Development and Technology, 6 (2001) 61–70. DOI: 10.1081/PDT-100000014
- Pikal M.J., Lukes A.L., Lang J.E., Gaines K., Quantitative crystallinity determinations for β-lactam antibiotics by solution calorimetry: correlations with stability, Journal of Pharmaceutical Sciences, 67 (1978) 767–773. DOI: 10.1002/jps.2600670609
- Pitt K.G., Webber R.J., Hill K.A., Dey D., Gamlen M.J., Compression prediction accuracy from small scale compaction studies to production presses, Powder Technology, 270 (2015) 490–493. DOI: 10.1016/j.powtec.2013.10.007
- Ponchel G., Montisci M.-J., Dembri A., Durrer C., Duchêne D., Mucoadhesion of colloidal particulate systems in the gastro-intestinal tract, European Journal of Pharmaceutics and Biopharmaceutics, 44 (1997) 25–31. DOI: 10.1016/S0939-6411(97)00098-2
- Price D.N., Kunda N.K., Muttil P., Challenges associated with the pulmonary delivery of therapeutic dry powders for preclinical testing, KONA Powder and Particle Journal, 36 (2019) 129–144. DOI: 10.14356/kona.2019008
- Roberts M., Ford J.L., Rowe P.H., Dyas A.M., MacLeod G.S., Fell J.T., Smith G.W., Effect of lubricant type and concentration on the punch tip adherence of model ibuprofen formulations, Journal of Pharmacy and Pharmacology, 56 (2004) 299–305. DOI: 10.1211/0022357022827
- Saleki-Gerhardt A., Zografi G., Non-isothermal and isothermal crystallization of sucrose from the amorphous state, Pharmaceutical Research, 11 (1994) 1166–1173. DOI: 10.1023/A:1018945117471
- Sasaki H., Karasawa K., Hironaka K., Tahara K., Tozuka Y., Takeuchi H., Retinal drug delivery using eyedrop preparations of poly-l-lysine-modified liposomes, European Journal of Pharmaceutics and Biopharmaceutics, 83 (2013) 364–369. DOI: 10.1016/j.ejpb.2012.10.014
- Seager H., Drug-delivery products and the Zydis fast-dissolving dosage form, Journal of Pharmacy and Pharmacology, 50 (2011) 375–382. DOI: 10.1111/j.2042-7158.1998.tb06876.x
- Sebhatu T., Ahlneck C., Alderborn G., The effect of moisture content on the compression and bond-formation properties of amorphous lactose particles, International Journal of Pharmaceutics, 146 (1997) 101–114. DOI: 10.1016/S0378-5173(96)04777-1
- Sebhatu T., Elamin A.A., Ahlneck C., Effect of moisture sorption on tabletting characteristics of spray dried (15 % amorphous) lactose, Pharmaceutical Research, 11 (1994) 1233–1238. DOI: 10.1023/A:1018973923831
- Sekiguchi K., Obi N., Studies on absorption of eutectic mixture. i. a comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man, Chemical & Pharmaceutical Bulletin, 9 (1961) 866–872. DOI: 10.1248/cpb.9.866
- Senior J.H., Fate and behavior of liposomes in vivo: a review of controlling factors, Critical Reviews in Therapeutic Drug Carrier Systems, 3 (1987) 123–193. PMID: 3542245.
- Shenfield G.M., Hill J.C., Infrequent response by diabetic rats to insulin-liposomes, Clinical and Experimental Pharmacology and Physiology, 9 (1982) 355–361. DOI: 10.1111/j.1440-1681.1982.tb00819.x
- Shimazawa M., Inoue Y., Masuda T., Onodera R., Tahara K., Shimizu Y., Mibe Y., Tsuruma K., Takeuchi H., Hara H., Topical diclofenac-loaded liposomes ameliorate laser-induced choroidal neovascularization in mice and non-human primates, Current Neurovascular Research, 14 (2017) 46–52. DOI: 10.2174/1567202614666161104115440
- Simonelli A.P., Mehta S.C., Higuchi W.I., Dissolution rates of high energy polyvinylpyrrolidone (PVP)-sulfathiazole coprecipitates, Journal of Pharmaceutical Sciences, 58 (1969) 538–549. DOI: 10.1002/jps.2600580503
- Sixsmith D., McCluskey D., The effect of punch tip geometry on powder movement during the tableting process, Journal of Pharmacy and Pharmacology, 33 (1981) 79–81. DOI: 10.1111/j.2042-7158.1981.tb13715.x
- Stubberud L., Arwidsson H.G., Hjortsberg V., Graffner C., Water-solid interactions. iii. effect of glass transition temperature, Tg, and processing on tensile strength of compacts of lactose and lactose/polyvinyl pyrrolidone, Pharmaceutical Development and Technology, 1 (1996) 195–204. DOI: 10.3109/10837459609029894
- Stubberud L., Forbes R.T., The use of gravimetry for the study of the effect of additives on the moisture-induced recrystallisation of amorphous lactose, International Journal of Pharmaceutics, 163 (1998) 145–156. DOI: 10.1016/S0378-5173(97)00382-7
- Sugihara H., Yamamoto H., Kawashima Y., Takeuchi H., Effectiveness of submicronized chitosan-coated liposomes in oral absorption of indomethacin, Journal of Liposome Research, 22 (2012) 72–79. DOI: 10.3109/08982104.2011.621128
- Sugimori K., Mori S., Kawashima Y., Characterization of die wall pressure to predict capping of flat- or convex-faced drug tablets of various sizes, Powder Technology, 58 (1989a) 259–264. DOI: 10.1016/0032-5910(89)80052-X
- Sugimori K., Mori S., Kawashima Y., Introduction of new index for the prediction of capping tendency of tablets, Chemical & Pharmaceutical Bulletin, 37 (1989b) 458–462. DOI: 10.1248/cpb.37.458
- Sugimoto M., Narisawa S., Matsubara K., Yoshino H., Nakano M., Handa T., Development of manufacturing method for rapidly disintegrating oral tablets using the crystalline transition of amorphous sucrose, International Journal of Pharmaceutics, 320 (2006) 71–78. DOI: 10.1016/j.ijpharm.2006.04.004
- Sun C.C., Dependence of ejection force on tableting speed—A compaction simulation study, Powder Technology, 279 (2015) 123–126. DOI: 10.1016/j.powtec.2015.04.004
- Sunamoto J., Iwamoto K., Takada M., Yuzuriha T., Katayama K., Polymer Coated Liposomes for Drug Delivery to Target Specific Organs, in: Anderson J.M., Kim S.W. (Eds.), Recent Advances in Drug Delivery Systems, Springer US, Boston, MA, 1984, pp. 153–162, ISBN: 978-1-4613-2745-5. DOI: 10.1007/978-1-4613-2745-5_10
- Tahara K., Nishikawa M., Matsui K., Hisazumi K., Onodera R., Tozuka Y., Takeuchi H., In vitro and in vivo characterization of drug nanoparticles prepared using PureNano™ continuous crystallizer to improve the bioavailability of poorly water soluble drugs, Pharmaceutical Research, 33 (2016) 2259– 2268. DOI: 10.1007/s11095-016-1964-7
- Takahashi T., Toyota H., Kuroiwa Y., Yoshino H., Kondou H., Yamashita K., Hakomori T., Takeuchi H., Quantitative evaluation of different rotary tablet presses by compaction velocity based on compaction simulation study, International Journal of Pharmaceutics, 558 (2019) 157–164. DOI: 10.1016/j.ijpharm.2018.12.032
- Takahashi T., Toyota H., Kuroiwa Y., Kondo H., Dohi M., Hakomori T., Nakamura M., Takeuchi H., Application of novel compaction indicator for the optimization of compaction conditions based on a compaction simulation study, International Journal of Pharmaceutics, 587 (2020) 119574. DOI: 10.1016/j.ijpharm.2020.119574
- Taketani Y., Usui T., Toyono T., Shima N., Yokoo S., Kimakura M., Yamagami S., Ohno S., Onodera R., Tahara K., Takeuchi H., Kuroda M., Topical use of angiopoietin-like protein 2 RNAi-loaded lipid nanoparticles suppresses corneal neovascularization, Molecular Therapy—Nucleic Acids, 5 (2016) e292. DOI: 10.1038/mtna.2016.1
- Takeuchi H., Chapter 2.15-Solubility and Dissolution Rate, in: Higashitani K., Makino H., Matsusaka S. (Eds.), Powder Technology Handbook (4th Edition), CRC Press, 2019, pp. 171–178, ISBN: 9781315184258. DOI: 10.1201/b22268
- Takeuchi H., Fuji Sylysia Chemical Ltd., Tablet and method for producing the same, Jpn. Patent No. 5026675 (2012).
- Takeuchi H., Handa T., Kawashima Y., Enhancement of the dissolution rate of a poorly water-soluble drug (tolbutamide) by a spray-drying solvent deposition method and disintegrants, Journal of Pharmacy and Pharmacology, 39 (1987a) 769–773. DOI: 10.1111/j.2042-7158.1987.tb05117.x
- Takeuchi H., Handa T., Kawashima Y., Spherical solid dispersion containing amorphous tolbutamide embedded in enteric coating polymers or colloidal silica prepared by spray-drying technique, Chemical & Pharmaceutical Bulletin, 35 (1987b) 3800–3806. DOI: 10.1248/cpb.35.3800
- Takeuchi H., Kojima H., Toyoda T., Yamamoto H., Hino T., Kawashima Y., Prolonged circulation time of doxorubicin-loaded liposomes coated with a modified polyvinyl alcohol after intravenous injection in rats, European Journal of Pharmaceutics and Biopharmaceutics, 48 (1999a) 123–129. DOI: 10.1016/S0939-6411(99)00029-6
- Takeuchi H., Kojima H., Yamamoto H., Kawashima Y., Enhanced tumour accumulation of doxorubicin with polymer-coated liposomes in rats, Pharmacy and Pharmacology Communications, 5 (1999b) 445–448. DOI: 10.1211/146080899128735144
- Takeuchi H., Kojima H., Yamamoto H., Kawashima Y., Polymer coating of liposomes with a modified polyvinyl alcohol and their systemic circulation and RES uptake in rats, Journal of Controlled Release, 68 (2000a) 195–205. DOI: 10.1016/S0168-3659(00)00260-1
- Takeuchi H., Kojima H., Yamamoto H., Kawashima Y., Evaluation of circulation profiles of liposomes coated with hydrophilic polymers having different molecular weights in rats, Journal of Controlled Release: official journal of the Controlled Release Society, 75 (2001a) 83–91. DOI: 10.1016/s0168-3659(01)00368-6
- Takeuchi H., Kojima H., Yamamoto H., Kawashima Y., Evaluation of the anti-tumour activity of liposomal doxorubicin coated with hydrophilic polymers in tumour-bearing mice, STP pharma sciences, 11 (2001b) 271–274.
- Takeuchi H., Kojima H., Yamamoto H., Kawashima Y., Passive targeting of doxorubicin with polymer coated liposomes in tumor bearing rats, Biological and Pharmaceutical Bulletin, 24 (2001c) 795–799. DOI: 10.1248/bpb.24.795
- Takeuchi H., Matsui Y., Sugihara H., Yamamoto H., Kawashima Y., Effectiveness of submicron-sized, chitosan–coated liposomes in oral administration of peptide drugs, International Journal of Pharmaceutics, 303 (2005a) 160–170. DOI: 10.1016/j.ijpharm.2005.06.028
- Takeuchi H., Matsui Y., Yamamoto H., Kawashima Y., Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats, Journal of Controlled Release, 86 (2003a) 235–242. DOI: 10.1016/S0168-3659(02)00411-X
- Takeuchi H., Nagira S., Aikawa M., Yamamoto H., Kawashima Y., Effect of lubrication on the compaction properties of pharmaceutical excipients as measured by die wall pressure, Journal of Drug Delivery Science and Technology, 15 (2005b) 177–182. DOI: 10.1016/S1773-2247(05)50023-6
- Takeuchi H., Nagira S., Yamamoto H., Kawashima Y., Design of solid dispersion particles of drug with fine porous carriers, Journal of the Society of Powder Technology, Japan, 40 (2003b) 157–162. DOI: 10.4164/sptj.40.157
- Takeuchi H., Nagira S., Yamamoto H., Kawashima Y., Solid dispersion particles of tolbutamide prepared with fine silica particles by the spray-drying method, Powder Technology, 141 (2004a) 187–195. DOI: 10.1016/j.powtec.2004.03.007
- Takeuchi H., Nagira S., Yamamoto H., Kawashima Y., Die wall pressure measurement for evaluation of compaction property of pharmaceutical materials, International Journal of Pharmaceutics, 274 (2004b) 131–138. DOI: 10.1016/j.ijpharm.2004.01.008
- Takeuchi H., Onodera R., Preparation of nano particles from a liquid phase and dosage form design, Funtai-Gijutsu, 7(6) (2015) 537–541.
- Takeuchi H., Sasaki H., Niwa T., Hino T., Kawashima Y., Uesugi K., Kayano M., Miyake Y., Preparation of powdered redispersible vitamin e acetate emulsion by spray-drying technique, Chemical & Pharmaceutical Bulletin, 39 (1991) 1528–1531. DOI: 10.1248/cpb.39.1528
- Takeuchi H., Thongborisute J., Matsui Y., Sugihara H., Yamamoto H., Kawashima Y., Novel mucoadhesion tests for polymers and polymer-coated particles to design optimal mucoadhesive drug delivery systems, Advanced Drug Delivery Reviews, 57 (2005c) 1583–1594. DOI: 10.1016/j.addr.2005.07.008
- Takeuchi H., Yamakawa R., Nishimatsu T., Takeuchi Y., Hayakawa K., Maruyama N., Design of rapidly disintegrating drug delivery films for oral doses with hydroxypropyl methylcellulose, Journal of Drug Delivery Science and Technology, 23 (2013) 471–475. DOI: 10.1016/S1773-2247(13)50068-2
- Takeuchi H., Yamamoto H., Kawashima Y., Mucoadhesive nanoparticulate systems for peptide drug delivery, Advanced Drug Delivery Reviews, 47 (2001d) 39–54. DOI: 10.1016/S0169-409X(00)00120-4
- Takeuchi H., Yamamoto H., Niwa T., Hino T., Kawashima Y., Mucoadhesion of polymer-coated liposomes to rat intestine in vitro, Chemical & Pharmaceutical Bulletin, 42 (1994) 1954–1956. DOI: 10.1248/cpb.42.1954
- Takeuchi H., Yamamoto H., Niwa T., Hino T., Kawashima Y., Enteral absorption of insulin in rats from mucoadhesive chitosan-coated liposomes, Pharmaceutical Research, 13 (1996) 896–901. DOI: 10.1023/A:1016009313548
- Takeuchi H., Yamamoto H., Toyoda T., Toyobuku H., Hino T., Kawashima Y., Physical stability of size controlled small unilameller liposomes coated with a modified polyvinyl alcohol, International Journal of Pharmaceutics, 164 (1998) 103–111. DOI: 10.1016/S0378-5173(97)00404-3
- Takeuchi H., Yasuji T., Hino T., Yamamoto H., Kawashima Y., Compaction Properties of Composite Particles Consisting of Lactose with Sodium Alginate Prepared by Spray-Drying, Pharmaceutical Research, 16 (1999c) 1193–1198. DOI: 10.1023/A:1018937227794
- Takeuchi H., Yasuji T., Yamamoto H., Kawashima Y., Temperature- and moisture-induced crystallization of amorphous lactose in composite particles with sodium alginate prepared by spray-drying, Pharmaceutical Development and Technology, 5 (2000b) 355–363. DOI: 10.1081/PDT-100100551
- Takeuchi H., Yasuji T., Yamamoto H., Kawashima Y., Temperature-induced crystallization and compactibility of spray dried composite particles composed of amorphous lactose and various types of water-soluble polymer, Chemical & Pharmaceutical Bulletin, 48 (2000c) 585–588. DOI: 10.1248/cpb.48.585
- Takeuchi Y., Ikeda N., Tahara K., Takeuchi H., Mechanical characteristics of orally disintegrating films: comparison of folding endurance and tensile properties, International Journal of Pharmaceutics, 589 (2020a) 119876. DOI: 10.1016/j.ijpharm.2020.119876
- Takeuchi Y., Murase Y., Tahara K., Takeuchi H., Impact of surface roughness of pre-treated punches and powder properties on prevention of sticking during pharmaceutical tableting, Journal of Drug Delivery Science and Technology, 60 (2020b) 101999. DOI: 10.1016/j.jddst.2020.101999
- Takeuchi Y., Nishimatsu T., Tahara K., Takeuchi H., Novel use of insoluble particles as disintegration enhancers for orally disintegrating films, Journal of Drug Delivery Science and Technology, 54 (2019) 101310. DOI: 10.1016/j.jddst.2019.101310
- Takeuchi Y., Umemura K., Tahara K., Takeuchi H., Formulation design of hydroxypropyl cellulose films for use as orally disintegrating dosage forms, Journal of Drug Delivery Science and Technology, 46 (2018) 93–100. DOI: 10.1016/J.JDDST.2018.05.002
- Tanimura S., Kawazoe H., Yamamoto H., Takeuchi H., Direct compaction of poorly compactable pharmaceutical powders with spray-dried HPC-L, Journal of the Society of Powder Technology, Japan, 43 (2006) 648–652. DOI: 10.4164/sptj.43.648
- Tanimura S., Tahara K., Takeuchi H., Spray-dried composite particles of erythritol and porous silica for orally disintegrating tablets prepared by direct tableting, Powder Technology, 286 (2015) 444–450. DOI: 10.1016/j.powtec.2015.08.011
- Tanino T., Aoki Y., Furuya Y., Sato K., Takeda T., Mizuta T., Occurrence of capping due to insufficient air escape during tablet compression and a method to prevent it, Chemical & Pharmaceutical Bulletin, 43 (1995) 1772–1779. DOI: 10.1248/cpb.43.1772
- Thongborisute J., Takeuchi H., Yamamoto H., Kawashima Y., Visualization of the penetrative and mucoadhesive properties of chitosan and chitosan-coated liposomes through the rat intestine, Journal of Liposome Research, 16 (2006) 127–141. DOI: 10.1080/08982100600680816
- Van der Voort Maarschalk K., Zuurman K., Vromans H., Bolhuis G.K., Lerk C.F., Stress relaxation of compacts produced from viscoelastic materials, International Journal of Pharmaceutics, 151 (1997) 27–34. DOI: 10.1016/S0378-5173(97)04889-8
- Vromans H., Bolhuis G.K., Lerk C.F., Kussendrager K.D., Bosch H., Studies on tableting properties of lactose. VI. Consolidation and compaction of spray dried amorphous lactose, Acta Pharmaceutica Suecica, 23 (1986) 231–240. PMID: 3799215.
- Waimer F., Krumme M., Danz P., Tenter U., Schmidt P.C., A novel method for the detection of sticking of tablets, Pharmaceutical Development and Technology, 4 (1999a) 359–367. DOI: 10.1081/PDT-100101371
- Waimer F., Krumme M., Danz P., Tenter U., Schmidt P.C., The influence of engravings on the sticking of tablets. investigations with an instrumented upper punch, Pharmaceutical Development and Technology, 4 (1999b) 369–375. DOI: 10.1081/PDT-100101372
- Werle M., Makhlof A., Takeuchi H., Carbopol-lectin conjugate coated liposomes for oral peptide delivery, Chemical and Pharmaceutical Bulletin, 58 (2010) 432–434. DOI: 10.1248/cpb.58.432
- Werle M., Takeuchi H., Chitosan-aprotinin coated liposomes for oral peptide delivery: development, characterisation and in vivo evaluation, International Journal of Pharmaceutics, 370 (2009a) 26–32. DOI: 10.1016/j.ijpharm.2008.11.013
- Werle M., Takeuchi H., Bernkop-Schnürch A., Modified chitosans for oral drug delivery, Journal of Pharmaceutical Sciences, 98 (2009b) 1643–1656. DOI: 10.1002/jps.21550
Author’s Short Biography
Hirofumi Takeuchi

Dr. Hirofumi Takeuchi is now Professor Emeritus and research Professor of Gifu Pharmaceutical University (2019–). He received his B.S. (1979), M.S. (1981) and Ph.D. (1984) in pharmacy and pharmaceutical sciences from Kyoto University. He joined Gifu Pharmaceutical University (GPU) in 1984, and worked as Lecture, Associate Professor and Professor of pharmaceutical engineering. He also worked as the dean of graduate school of GPU (2010–2011), dean of the faculty of GPU (2012–2013) and vice president of GPU (2014). He became the research Professor of Advanced Pharmaceutical Process Engineering (APPEN) as well as Professor Emeritus of GPU after retirement in 2019.
 https://ror.org/0372t5741
https://ror.org/0372t5741 


;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/39_2022017_28.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)