Abstract
Several bacterial pathogens contain membrane ligands that facilitate their binding and internalization into human tissues. In this study, lipooligosaccharides (LOS) from the respiratory pathogen non-typeable Haemophilus influenzae (NTHi) were isolated from the bacterial surface and evaluated as a nanoparticle coating material to facilitate uptake into the respiratory epithelium. NTHi clinical isolates were screened to select a strain with high binding potential due to their elevated phosphorylcholine content. The association of particles with human bronchial epithelial cells was investigated as a function of particle surface chemistry and incubation time, and the uptake mechanism was evaluated via chemical inhibitor and receptor activation studies. A more than two-fold enhancement in particle uptake was achieved by coating the particles with LOS compared to uncoated or gelatin-coated particles, which was further increased by activating the platelet-activating factor receptor (PAFR). These findings demonstrate that bacterial-derived LOS ligands can enhance the targeting and binding of nanoparticles to lung epithelial cells.
1. Introduction
Targeted drug delivery is a powerful strategy to treat diseases that affect human tissues, offering high drug concentrations at the target site with limited exposure of other tissues. Efficient delivery can be achieved using drug-loaded nanoparticles with chemical and physical properties tuned to avoid host defense mechanisms and inhibit nonspecific distribution (Fenton et al., 2018; Patra et al., 2018). Active targeting can further enhance delivery to specific target organs or cells within the body. With appropriate surface functionality, such as through the attachment of ligands that recognize receptors on cellular surfaces, particulate carriers show improved efficiency in penetrating cells in vitro. One challenge in achieving active targeting has been the identification of cellular targets, which is limited by our knowledge of disease-specific cell receptors and their ligands. A potentially useful class of ligands is produced and used by human pathogenic organisms to infect the respiratory tract.
Haemophilus influenzae (H. influenzae), a Gram-negative bacterium that colonizes the nasopharynx of up to 80 % of humans, is a leading cause for bacterial meningitis and other widespread bacteremic diseases (Cochi and Broome, 1986; Foxwell et al., 1998). While a polysaccharide conjugate vaccine is available for and is effective against H. influenzae serotype b, the vaccine is not effective against the subtype that lacks a capsule, called non-typeable H. influenzae (NTHi) (St. Geme, 1996). NTHi causes localized opportunistic infections including otitis media, sinusitis, conjunctivitis, bronchitis and pneumonia (Turk, 1984). It can infect cystic fibrosis (CF) patients early in childhood and prime the lungs for later infection with the bacterium Pseudomonas aeruginosa, which is correlated with higher morbidity and mortality in CF patients (Starner et al., 2006).
Diseases due to NTHi often start with colonization of the respiratory epithelium (Turk, 1984). The surface of NTHi is decorated with lipooligosaccharides (LOS) which contain a lipid structure that embeds in the outer leaf of the bacterial outer membrane and an oligosaccharide structure that facilitates bacterial pathogenesis. By expressing specific carbohydrates, such as phosphorylcholine (ChoP), as a terminal structure on the LOS, the bacteria are imbued with two important defenses. First, ChoP expression reduces the ability of LOS-specific or bactericidal antibodies to bind to the bacteria through molecular mimicry of host structures, enabling the bacteria to evade the host immune system and limit their pulmonary clearance (Clark et al., 2012; Pang et al., 2008; Weiser et al., 1998). Second, the presence of ChoP on the NTHi surface enhances bacterial adherence to host cells by binding to the platelet-activating factor receptor (PAFR), effectively reducing mucociliary clearance of the bacteria and enhancing bacterial invasion of the respiratory epithelium (Swords et al., 2000, 2001; Turk, 1984). The effectiveness of OS in inducing cellular uptake is dependent on the specific LOS glycoform, with LOS containing higher ChoP content resulting in enhanced NTHi colonization and persistence in the lungs (Swords et al., 2000; Weiser et al., 1998). These findings suggest that isolated ChoP-containing LOS could be a potential targeting ligand for drug delivery particles to mimic bacterial entry into lung cells and thus deliver drugs directly to infected lung cells.
We hypothesized that LOS ligands naturally expressed on bacterial cell surfaces from NTHi could be utilized as targeting ligands to facilitate particle attachment and uptake into lung epithelial cells. The expression of ChoP on several clinical isolates of NTHi was measured and an NTHi strain with high ChoP content chosen for LOS isolation. Nanoparticle uptake was evaluated in 16HBEo-bronchial epithelial cells in the absence and presence of endocytosis pathway inhibitors and activators of PAFR.
2. Experimental
2.1 Culture of NTHi bacteria
NTHi clinical strains 2019 and 3198, isolated from patients with chronic obstructive pulmonary disease (Campagnari et al., 1987), and strain 956 were reconstituted from frozen glycerol stocks and propagated on chocolate agar (a non-selective, enriched growth medium) or brain heart infusion (BHI) agar (selective medium for NTHi) supplemented with 10 μg/mL hemin and 1 μg/mL nicotinamide adenine dinucleotide (NAD) at 37 °C and 5 % CO2. All culture chemicals were obtained from Sigma-Aldrich.
2.2 Detection of ChoP on NTHi cells via ELISA and dot immunoblot
The three NTHi strains were screened by enzyme-linked immunosorbent assay (ELISA) to identify bacterial strains and growth conditions that result in bacteria with high ChoP activity. An NTHi cell suspension (8.0 × 107 cells/mL) was added to each well of a 96-well plate (Corning Inc. product 3590; clear, flat-bottom polystyrene, high-binding). The plate was dried in an oven uncovered overnight at 40 °C. The wells were then washed three times with 200 μL ELISA wash solution (0.98 % w/v sodium acetate, 0.9 % w/v NaCl, 0.05 % v/v Tween 20 in purified water from NanoPure® Infinity UV Barnstead system). The primary monoclonal antibody 12D9, which was raised directly against ChoP, was diluted 1:25 and added to each well. The plate was incubated overnight at room temperature, then washed. Alkaline phosphatase-conjugated goat anti-mouse IgG AffiniPure F(ab’)2 (Jackson ImmunoResearch Inc.) diluted 1:2000 in ELISA buffer was added to each well and incubated for 1 h, after which the wells were again washed. The substrate, nitrophenyl phosphate bis (cyclohexyl ammonium) (pNPP), was prepared at 1 mg/mL in diethanolamine buffer (10 % w/v diethanolamine and 0.01 % w/v MgCL2 in purified water, pH 9.8) immediately before use, added to all wells, and incubated in the dark. The substrate was then hydrolyzed by alkaline phosphatase to develop a yellow water-soluble product, p-nitrophenol. The relative absorbance of p-nitrophenol at 405 nm was read using a SpectraMax® M2 spectrophotometer (Molecular Devices) after 2 h incubation. Wells with no cells, no primary antibody, or no secondary antibody served as negative controls (n = 4). Dot immunoblot confirmed the relative high ChoP expression on NTHi 3198 cell surfaces.
2.3 Proteinase K/hot phenol method to isolate LOS from NTHi bacterial cells
Due to its relatively high ChoP expression, LOS was isolated from NTHi strain 3198 using the proteinase K digest, phenol-water extraction method (Apicella, 2008; Galanos et al., 1969). NTHi was cultured on supplemented BHI agar overnight, harvested, digested in ELISA buffer containing 1 % w/v sodium dodecyl sulfate (SDS, Sigma-Aldrich®) and 50 μg/mL proteinase K at 65 °C for 1 h, and incubated at 37 °C overnight to lyse the cells and digest the proteins. The mixture was washed three times via precipitation in 3 M sodium acetate and cold absolute ethanol overnight at −20 °C, then centrifuged at 3200 × g at 4 °C (Eppendorf centrifuge I810R). The precipitate was washed with ethanol to remove residual SDS. After the final precipitation, the pellets were air-dried at room temperature for 2 h and then suspended in 15 mL of DNase I buffer (10 mM Tris-HCl, 2.5 mM MgCl2, 0.5 mM CaCl2 in purified water, pH 7.6). Solutions of 10 μg/mL DNase (Roche) and 10 μg/mL RNase (Qiagen) were added and the solution incubated at 37 °C in a water bath for 2 h to digest residual nucleic acids. Finally, a phenol extraction of the LOS nuclease-treated mixture removed residual protein contamination. The LOS mixture was isolated from phenol soluble components using an equal volume of pre-heated phenol and incubated at 65 °C for 20 min, then on ice for 1 h. Upon cooling, the suspension was separated into a phenol phase containing proteins, an interphase containing cell-wall material, and an aqueous phase enriched in LOS. The top aqueous layer was collected and washed three times before lyophilization and storage at 4 °C in a desiccant box. The activity of the isolated LOS was confirmed using the colorimetric ELISA described above, except the primary antibody 12D9 was prepared at 1:50 dilution in ELISA buffer, the secondary antibody was added at 1:2000 dilution in ELISA buffer, and the time for p-nitrophenol to develop was 3 h.
2.4 Preparation of LOS-functionalized particles
Polystyrene particles (0.2 μm plain white Polybead® or Fluoresbrite® polystyrene particles, Polysciences Inc.) were prepared for functionalization by washing twice with carbonate buffer (15.9 mg Na2CO3 and 25.9 mg NaHCO3 in 100 mL purified water, pH 9.6) an centrifuging at 12,000 × g for 30 min, followed by resuspension at a concentration of 2 × 109 particles/mL. Particles with a nominal size of 0.2 μm were chosen to match the mean width of an NTHi bacterium.
To achieve particle functionalization, polystyrene particles were added to a solution containing NTHi 3198 LOS at either a low (5 ng/mL) or high (5 μg/mL) ligand concentration, mixed on a rotator for 2 h, washed twice with carbonate buffer, and resuspended in carbonate buffer. A subset of uncoated and high ligand coated polystyrene particles were exposed to a protein mixture of 0.1 % gelatin (Type B from bovine skin derived from lime-cured tissue, average MW 50,000–100,000 g/mol; Sigma G9382) solution in carbonate buffer. Particles were either stored in carbonate buffer at a final concentration of 2 × 109 particles/mL at 4 °C (cell association studies) or lyophilized overnight in a Labconco® FreeZone® 4.5-liter freeze dry system at a chamber pressure <0.02 mbar and collector temperature at or below −50 °C.
2.5 Particle characterization
Lyophilized polystyrene particles were prepared for size and zeta potential measurements by dispersion in water or serum-free medium (pH 7.4) via three cycles of vortexing for 90 s and sonication for 10 min in cold water bath at 4 °C. Particle suspensions were placed into folded capillary cells (DTS 1060C cuvettes, Malvern Instruments) and the average particle hydrodynamic diameter and zeta potential (n = 3) measured using a Zetasizer® Nano ZS (Malvern Panalytical LTD.). Particle size and morphology were confirmed via scanning electron microscopy using an S-4800 Field Emission Scanning Electron Microscope (FE-SEM). Lyophilized particles were tapped onto SEM stubs coated with double-sided black carbon tape and sputter coated with Au/Pd at 10 mA and an pressure above 7 × 10−2 mbar for 3 min (K550 Emitech® sputter coater).
To prepare particles for surface chemical analysis, washed particles were dispersed in water, and 20 μL of the suspension was added and dried on clean silicon wafers. Chemical analysis was carried out using a Kratos® Axis Ultra X-ray photoelectron spectrometer (XPS) with concentric hemispherical electron energy analyzers combined with an established delay-line detector. The incident radiation from monochromatic Al Kα X-rays (1486.6 eV) at 150 W (accelerating voltage 15 kV, emission current 10 mA) was projected 45° to the sample surface and the photoelectron data were collected at a takeoff angle of θ = 90°. The base pressure in the analysis chamber was maintained at 1.0 × 10−9 torr. Survey scans were taken at a pass energy of 160 eV and carried out over a 1200 eV ~ −5 eV binding energy range with 1.0 eV steps and a dwell time of 200 ms. High resolution scans of C 1s, O 1s, N 1s, P 2p, and S 2p were taken at a pass energy of 20 eV with 0.1 eV steps and a dwell time of 2000 ms. Spectral analysis was conducted using CasaXPS software (version 2.3.17dev6.4k). Spectra were calibrated using the adventitious carbon C 1s peak at 285.0 eV.
2.6 Isothermal titration calorimetry (ITC)
ITC measures the heat released or absorbed during a biomolecular binding event, enabling accurate determination of the enthalpy of binding and the stoichiometry of each binding event at different concentrations of ligand, including proteins on nanoparticles. The GE MicroCal® iTC200 (GE Healthcare) was used in this study. The accuracy of the instrument was verified by titrating a 5 mM standard CaCl2 solution into a 0.4 mM standard EDTA solution, both provided by the manufacturer. LOS and nanoparticle solutions were incubated in a 25 °C water bath for 15 min prior to use to ensure temperature equilibration. LOS solution at a concentration of either 20, 30, 50, or 100 μg/mL was loaded into the syringe and 625 μg/mL nanoparticle solution (the mass of nanoparticles in the original particle suspension was provided by the manufacturer) was loaded into the sample cell, then the system was equilibrated to 25 °C. Experimental parameters were set as follows: total number of injections = 20 (one 0.4 or 0.5 μL injection followed by nineteen 2 μL injections); cell temperature = 25 °C; initial delay = 60–200 s; syringe concentration = variable; mixing speed = 700 rpm. Accounting for the dilution in the ITC cell, the 30 and 50 μg/mL injections were equivalent to a final LOS concentration of about 4 and 7 μg/mL, respectively, interacting with the nanoparticles. Controls included carbonate buffer titrated into 625 μg/mL of nanoparticles and 50 μg/mL LOS titrated into carbonate buffer (to determine heat of dilution). Origin 7.0 software was used to convert all raw ITC data into DeltaH plots, and its regression function was used to calculate the stoichiometry, enthalpy, entropy and dissociation constant (Kd) of each titration.
2.7 Cultivation of immortalized bronchial epithelial cells
The 16HBE14o- cell line, generated by transformation of normal bronchial cells obtained from a heart–lung transplant patient, was generously provided by Professor Gruenert, Department of Otolaryngology, University of California, San Francisco (Gruenert et al., 1988). Cells were propagated in cell culture medium composed of Eagle’s minimum essential medium with Earle’s salts (Life Technologies Co.) supplemented with 10 % fetal bovine serum (FBS, Atlanta Biologics Inc.), 2 mM L-glutamine (Life Technologies Co.), 100 μg/mL streptomycin and 100 units/mL penicillin G (Life Technologies Co.) in T-75 flasks (Greiner Bio-One). The cell culture medium was replaced every other day. At 80 % cell coverage (4–6 days), the cells were passaged by washing three times with biological buffer (prepared as a 10-times concentrated solution containing 3.795 g NaCl, 0.35 g Na2HPO4, 0.20 g KCl, 0.99 g glucose, and 1.265 g HEPES in 1 L of purified water, pH 7.4), detachment by treatment with Trypsin/EDTA (Life Technologies Co.) for 5–10 min at 37 °C and 5 % CO2 with gentle agitation, and addition of the cell suspension to new flasks with 12 mL of media per flask. Passages 20 to 40 were used.
2.8 Immunofluorescent detection of PAFR on 16HBEo- cells
The expression of PAFR on the surface of 16HBEo-cells was determined using flow cytometry and confocal microscopy. Cells were prepared for flow cytometry by suspending cells in biological buffer containing 0.2 % Tween 20, centrifuging at 300 × g for 10 min, then incubating in cold methanol (−20 °C) for 10 min at −20 °C. The fixed cells were washed with biological buffer containing 0.2 % Tween 20 (Sigma) and incubated with 10 % normal goat serum in biological buffer containing 0.2 % Tween 20 at 4 °C overnight. Cells were then centrifuged, redispersed in biological buffer at a concentration of 1 × 107 cells/mL. To stain for PAFR expression, cells were incubated with 50 μL of 8 μg/mL solution of IgG2a PAFR (human) monoclonal antibody raised in mouse (product 160600, Cayman Chemical) in biological buffer for 30 min at room temperature. The controls were no stain, secondary antibody control, and isotype control using 8 μg/mL solution of mouse IgG2ak clone eBM2a (eBiosciencesTM) in biological buffer. Cells were then washed six times with cold biological buffer containing 0.2 % Tween 20 and incubated with 50 μL of either 6 % normal goat serum in biological buffer (no stain control) or 0.1 μg/mL solution of Alexa Fluor 647 Goat Anti-Mouse IgG (H+L) antibody (Life Technologies Co.) in biological buffer for 15 min. Cells were again washed six times with cold biological buffer containing 0.2 % Tween 20. After antibody treatment, cell suspensions were analyzed immediately using an LSR II flow cytometer (BDTM Biosciences).
To confirm and visualize PAFR expression, cells were cultured onto collagen-coated cover slips in 24-well plates and incubated overnight. The cells were washed three times with biological buffer containing 0.2 % Tween 20. Immunostaining of the cell membranes was conducted as described above, except 100 μL of each antibody solution was added to the cells and the secondary antibody Alexa Fluor 568 Goat Anti-Mouse IgG (H+L) antibody (Life Technologies Co.) was added at a concentration of 4 μg/mL. After cell washing, the glass coverslip containing the cell membrane was mounted to a microscope slide using antifade reagent with the nuclear stain DAPI, then kept at 4 °C until imaged. Cell layers were imaged using a Zeiss LSM 710 confocal microscope with 40× oil immersion objective lens.
2.9 Particle association with bronchial epithelial cells
To prepare cells for particle binding and uptake studies, 16HBEo- cells were propagated on collagen-coated glass coverslips. A volume of 100 μL of cells was dispensed, at 2 × 105 cells/well, onto dried collagen-coated coverslips and incubated one day until reaching 80 % confluency, as determined by visual inspection. Suspensions of fluorescent yellow particles with or without coating (0.22 μg/mL, 100 μL/well) were added to cells and incubated for 0.5, 2, 4, 18, or 24 h (n = 4 wells of cells per treatment) to quantify uptake with time. In each case, after incubation the cells were washed three times to remove unbound particles.
The number of cells associated with particles was quantified using an LSR II flow cytometer (BD Biosciences) with a 488 nm excitation source. Cells were processed for flow cytometry by incubating the cells with enzyme-free cell dissociation buffer (Life Technologies Co.) for 0.5–1 h at room temperature, centrifuging at 300 × g for 10 min at 4 °C, then dispersing in cold biological buffer by gentle pipetting to obtain a single cell suspension. Raw data were processed to gate the live cell population(s) based on cell size (forward scatter) and cell granularity (side scatter), effectively removing dead cells, cell debris and particle agglomerates. Gating was confirmed with controls, i.e. cells not treated with particles, particles alone, and cells and particles mixed briefly together. The population of cells containing particles were identified based on fluorescence intensity using the Probability Binning (PB) Chi(T) value (Roederer et al., 2001). The percentage of cells in the population was quantified using the Enhanced Normalization Subtraction (ENS) method in FlowJo® (Bagwell, 1996). Results were collected for at least 10,000 cells (n = 4).
Confocal microscopy was used to assess the location of particles within the cell cultures. After particle treatment, cell membranes were washed three times to remove unbound particles and secretions produced during the incubation period. Cell membranes were then stained with 1 μg/mL DiI D-282 (Life Technologies Co.), a lipophilic indocarbocyanine fluorescent red dye (549 nm/565 nm), in cell culture medium for 30 min, washed with biological buffer three times to remove excess dye, fixed with 2 % w/v paraformaldehyde for 30 min, blocked with 50 mM ammonia chloride for 10 min, and washed with biological buffer once to remove excess reagents. Triton X-100 prepared at 0.05 % w/v in biological buffer was used to permeabilize the cell membrane for 10 min. The membranes were washed, the cover glass containing the membranes mounted to a microscope slide, and the cells exposed to ProLong® Gold antifade reagent with the blue-fluorescent nuclear 4’,6-diamidino-2-phenylindole (DAPI 358 nm/461 nm; Life Technologies Co.) to stain the cell nuclei. The cells were kept at 4 °C until imaged. Z-stacks of cell layers were obtained using a LSM 710 upright confocal microscopy (Zeiss). A 40× oil immersion objective lens was used to take images with step size of 200 nm. Images were rendered in MATLAB.
2.10 Cell uptake inhibitor and activator studies
The toxicity of inhibitors to 16HBEo- cells was first determined using the MTS assay (CellTiter 96® Aqueous One Solution Cell Proliferation Assay; Promega Co.). Cells were seeded onto 96-well plates at 3 × 104 cells/well, incubated for 24 h, then treated for 0–5 h with one of the following inhibitors: 0.1 % NaN3 with 50 mM 2-deoxyglucose, 5 μg/mL cytochalasin D, 80 μM dynasore, 200 μM genistein, 10 μg/mL chlorpromazine, or 5 mM methyl β-cyclodextrin. Negative and positive controls were cells treated with biological buffer (absorbance normalized to 100 % cell viability) and cells treated with 2 % w/v SDS, respectively. Based on toxicity results, uptake studies in the presence of inhibitors were limited to time points less than 3 h, and studies with inhibitors of clathrin-mediated endocytosis were not conducted as cell viability with these inhibitors was 40–95 % at all time points.
To determine the mechanisms by which particles were taken up by cells, 16HBEo- cells were pre-treated with one of the following inhibitors one hour prior to particle treatment: 0.1 % NaN3 with 50 mM 2-deoxyglucose, 5 μg/mL cytochalasin D, 80 μM Dynasore, 200 μM genistein, or 5 mM methyl β-cyclodextrin. The receptor PAFR was activated by pre-treating cells with PAF (10 μM) for 2 h. The treatment solutions were removed then replaced with the particle suspension containing fresh inhibitor or PAF and incubated. After incubation, cells were washed three times to remove unbound particles and secretions produced during the incubation period. Particle–cell association was measured by flow cytometry.
2.11 Statistical analysis
Significance (p < 0.05) of differences was determined by one-way ANOVA with Holm-Sidak’s multiple comparison tests using GraphPad Prism software (version 6.05).
3. Results
3.1 Selection and characterization of NTHi bacteria
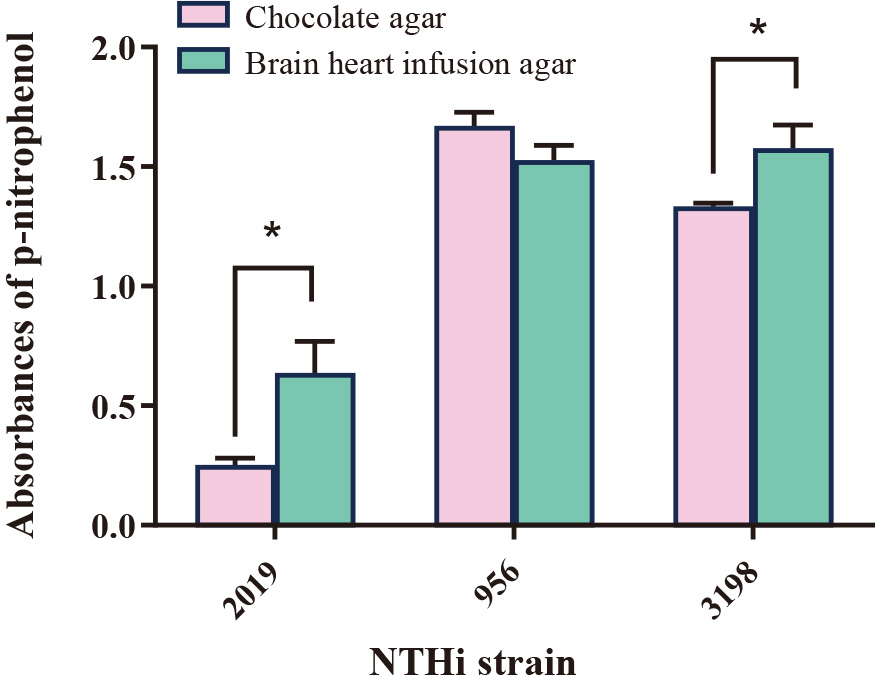
Since ChoP content is vital for epithelial cell binding, LOS with high ChoP content may enhance association with cells containing PAFR. Three NTHi strains were screened to select for the strain with a high activity of ChoP. NTHi strains 956 and 3198 expressed significantly higher ChoP activity, as measured by absorbance of p-nitrophenol, than the 2019 strain (Fig. 1). The higher level of ChoP activity on NTHi 3198 compared to NTHi 2019 was confirmed by dot immunoblot, with strain 3198 expressing more ChoP on its surface at shorter exposure times (1 s) than strain 2019 (30 s), as observed in the higher intensity and positive stained area of dark spots. This is in agreement with published literature that reports 78 % of strain 3198 expresses ChoP with high reactivity, compared to 9 % of strain 2019 (Swords et al., 2000; Weiser et al., 1997). Further, the expression of ChoP on strains 2019 and 3198 was higher when bacteria were cultured on BHI agar than on chocolate agar; in contrast, no statistical difference in ChoP expression was observed for strain 956 grown on the two substrates (Fig. 1). Strain 3198 cultured on supplemented BHI agar medium was chosen for LOS isolation and coating onto particles.
Electron microscopy images of the uncoated polystyrene particles confirmed that particles were spherical, with smooth surfaces (Fig. 2(a)). Upon particle exposure to carbonate buffer, salt crystals were observed attached to the particle surfaces (Fig. 2(b)). Coating the polystyrene particles with 5 ng/mL LOS did not change their appearance compared to the carbonate buffer-washed particles (Fig. 2(c)), as salt crystals remained attached to the particle surface; however, upon coating with the higher concentration of LOS (5 μg/mL), salt crystals were no longer observed, suggesting LOS displaced the salt on the particle surface (Fig. 2(d)). Similarly, exposing uncoated particles with a protein solution (gelatin) resulted in visible residue on the particle surface (Fig. 2(f)); however, this residue was not observed on particles that were pre-coated with 5 μg/mL LOS prior to gelatin coating (Fig. 2(e)).

When suspended in water, uncoated particles exhibited low polydispersity with a hydrodynamic diameter of ~255 nm, and a zeta potential of about −50 mV (Table 1). When placed in carbonate buffer, the particles agglomerated slightly. Agglomeration was maintained with LOS coating and was intensified with gelatin coating. When placed in serum-free media for cell studies, particles generally de-agglomerated, except for the 5 μg/mL LOS-coated particles which exhibited slightly higher agglomeration. In addition, particles suspended in media exhibited more neutral zeta potentials than the same particles in water. Serum-free media contains various salts that likely compressed the electrical double layer surrounding the particles, resulting in a reduction in the zeta potential. Particles coated with 5 μg/mL LOS exhibited the highest zeta potential in media at about −25 mV, suggesting significant surface coverage with the negatively charged LOS. LOS molecules have COOH groups on their carbohydrate structures which are negatively charged at physiologic pH, in addition to the ChoP moieties which are zwitterionic at physiologic pH.
Table 1
Characterization of geometric size and zeta potential of polystyrene particles (200 nm nominal diameter) with various surface coatings in water and serum-free media. Values are reported as mean ± SD (n = 3).
| Particle coating |
Suspension in water |
Suspension in serum-free media |
| Particle diameter (nm) |
Zeta potential (mV) |
Particle diameter (nm) |
Zeta potential (mV) |
| Uncoated |
255.15 ± 6.6 |
−50.0 ± 5.3 |
— |
— |
| Carbonate buffer |
381.0 ± 14.2 |
−40.9 ± 0.7 |
157.7 ± 9.9 |
–10.4 ± 1.2 |
| 5 ng/mL LOS |
295.2 ± 6.4 |
−46.8 ± 2.2 |
183.7 ± 7.4 |
–8.8 ± 0.5 |
| 5 μg/mL LOS |
351.6 ± 14.8 |
−47.8 ± 2.1 |
461.1 ± 7.8 |
–24.5 ± 2.2 |
| 5 μg/mL LOS and gelatin |
555.4 ± 254.6 |
−39.5 ± 1.1 |
187.7 ± 13.8 |
–7.6 ± 0.9 |
| Gelatin |
1053.8 ± 372.5 |
−36.4 ± 4.9 |
165.7 ± 3.2 |
–7.3 ± 0.9 |
The surface chemistry of uncoated and functionalized particles was investigated by XPS (Table 2). To confirm the existence of a coating on the functionalized particle surfaces, the raw coating materials were also analyzed. LOS contained mostly carbon (49.6 %) and oxygen (35.2 %), with the remaining composed of 9.3 % nitrogen, 5.6 % phosphate, and 0.3 % sulphur. Gelatin contained no phosphate, but was comprised of 65.8 % carbon, 18.7 % oxygen, 15 % nitrogen and 0.2 % sulfur. Gelatin also had a higher C/O ratio at 3.5, compared to LOS at 1.4. In contrast, the uncoated particle surface contained carbon, oxygen, and sulfur since the polystyrene particles used in this study had a polystyrene backbone composed of carbon and oxygen, and sulfate function groups. The polystyrene particles had the smallest C/O ratio at 0.36. Some residual phosphate (7 %) was also observed on the uncoated particle surface, likely due to contamination from the water washes. Given these differences among the raw materials, the phosphate, nitrogen, and C/O ratio were used to confirm functionalization of the particles.
Table 2
Surface elemental analysis of LOS, gelatin, and polystyrene particles with various surface coatings via X-ray photoelectron spectroscopy.
| Material |
% Atomic concentration |
C/O ratio |
| C 1s |
O 1s |
N 1s |
P 2p |
S 2p |
| LOS |
|
49.6 |
35.2 |
9.3 |
5.6 |
0.3 |
1.4 |
| Gelatin |
|
65.8 |
18.7 |
15.3 |
0.0 |
0.2 |
3.5 |
| Particles |
Uncoated |
19.5 |
54.3 |
0.0 |
6.7 |
19.5 |
0.36 |
|
5 ng/mL LOS |
38.6 |
40.8 |
1.0 |
5.0 |
14.6 |
0.9 |
|
5 μg/mL LOS |
57.7 |
29.2 |
0.5 |
3.1 |
9.6 |
2.0 |
|
5 μg/mL LOS and gelatin |
62.7 |
24.9 |
4.1 |
2.4 |
5.9 |
2.5 |
|
Gelatin |
75.9 |
16.7 |
4.5 |
0.6 |
2.3 |
4.5 |
Particles coated with a 5 ng/mL LOS solution exhibited an elemental surface indicative of a mostly uncoated polystyrene surface with a small amount of LOS adsorbed (C/O ratio of 0.9, between that of LOS and polystyrene; with existence of N indicating some LOS adsorption). This suggests that LOS was on the particle surface but did not completely coat the entire surface. With a higher LOS coating solution concentration (5 μg/mL), the C/O ratio increased to 2.0, suggesting a higher concentration of LOS on the surface. In addition, the phosphorus and sulphur concentrations at the surface decreased to 3.1 % and 9.6 %, respectively, suggesting that these moieties were buried beneath the carbon and oxygen backbone of the LOS. When uncoated particles or LOS-coated particles were coated with gelatin, the nitrogen concentration and C/O ratio increased significantly compared to their uncoated counterparts, to values closer to that of pure gelatin. This suggests that gelatin partially masked the surface of the particles, even in the presence of LOS.
The LOS coating process was further analyzed by ITC to determine the stoichiometry of LOS binding to the polystyrene particles. ITC experiments were carried out using four different concentrations of LOS (Fig. 3). The regression lines for the 20 and 100 μg/mL titrations exhibited negligible slope and therefore not analyzed. For the 30 and 50 μg/mL titrations, the amount of LOS bound to the particle surface increased with an increase in LOS concentration resulting in an estimated 17.7 LOS binding sites per particle (titration with 30 μg/mL LOS) and 33.9 LOS binding sites per particle (titration with 50 μg/mL LOS). Enthalpies of binding were large and negative for all titrations (−2 kcal/mol to −10 kcal/mol), indicating strong binding of LOS to the particle surface.
3.3 Uptake of particles in 16HBEo- lung epithelial cells
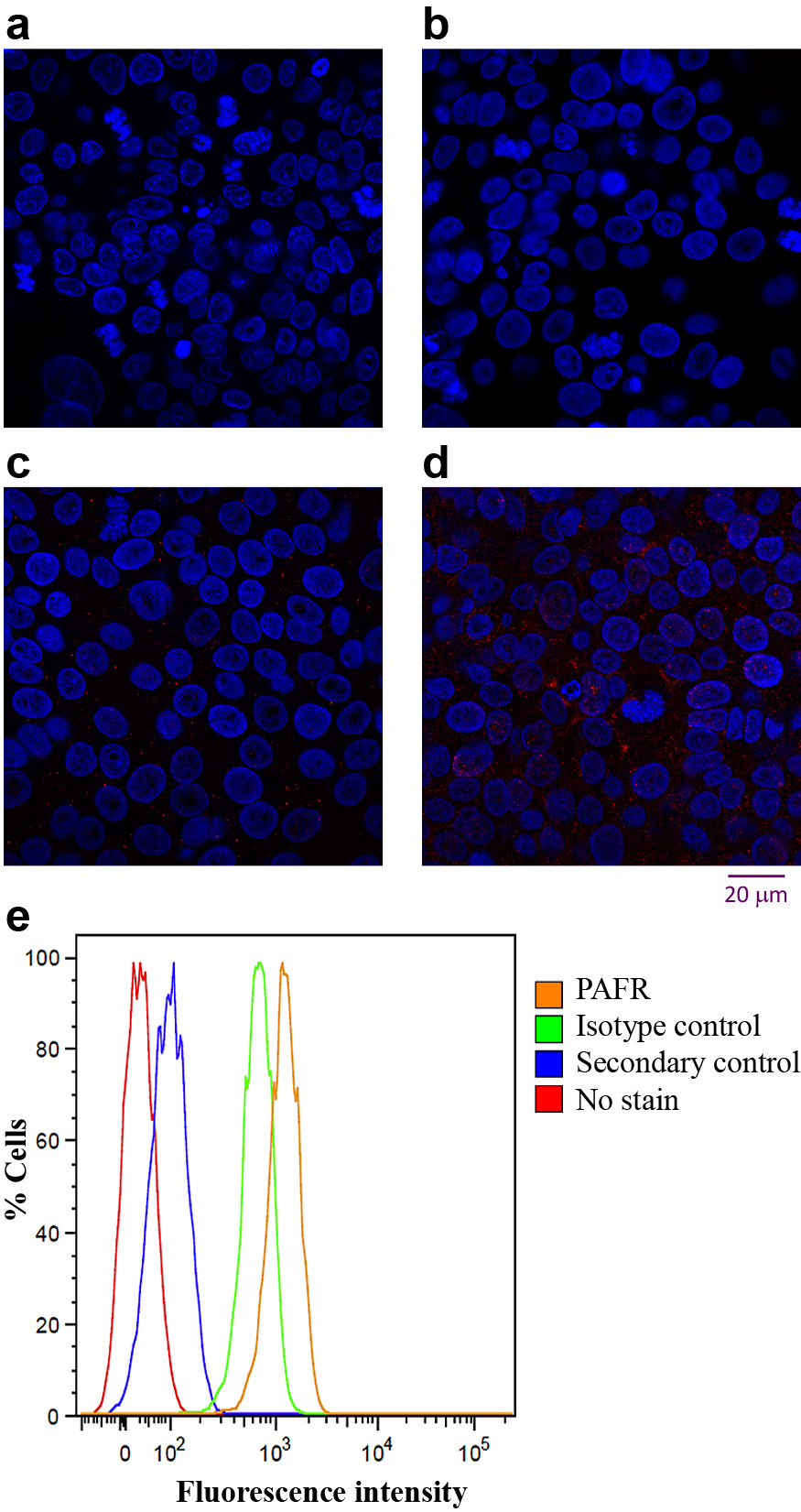
PAFR expression in 16HBE14o- cells was confirmed via immunostaining (Fig. 4). The secondary and isotype controls exhibited low levels of staining due to nonspecific receptor interactions (Figs. 4(b) and 4(c), respectively), which was quantified by flow cytometry via fluorescence intensity (Fig. 4(e)). PAFR expression on the cell surface was observed as diffuse staining across the entire cell surface (Fig. 4(d)). Flow cytometry confirmed that the entire population of 16HBEo- cells stained for PAFR exhibited higher intensity of fluorescence than controls, confirming the cells expressed PAFR (Fig. 4(e)).
Next, lung epithelial cells were exposed to uncoated, gelatin-coated, or functionalized particles for up to 24 h and the percentage of cells associated with particles quantified by flow cytometry. Uncoated particles and particles coated with 5 ng/mL LOS behaved similarly, with little uptake from 0–19 h (<10 % of cells were associated with particles) followed by a small increase at 24 h to 10 % (Fig. 5(a)). Gelatin coated particles were associated with at most 12 % of cells at 4 h, which was not statistically different than uncoated particles. However, particles coated with 5 μg/mL LOS were associated with about 12 % of cells at time 0 and this rose steadily to about 35 % at 24 h. Confocal microscopy confirmed that the 5 μg/mL LOS-coated particles were internalized into the 16HBEo-cells and not just attached to the cell surface (Fig. 5(b)).

The energy dependence of particle uptake in 16HBEo- cells was quantified after cell incubation with NaN3/2-deoxyglucose, which inhibits all energy-dependent pathways (Fig. 6). Incubation of cells with NaN3/2-deoxyglucose resulted in complete loss of uptake of the 5 μg/mL LOS-coated particles, suggesting these particles are taken up by endocytosis. No other particle type experienced inhibited cell uptake after cell exposure to NaN3/2-deoxyglucose and thus may be taken up by both energy-dependent and energy-independent pathways.
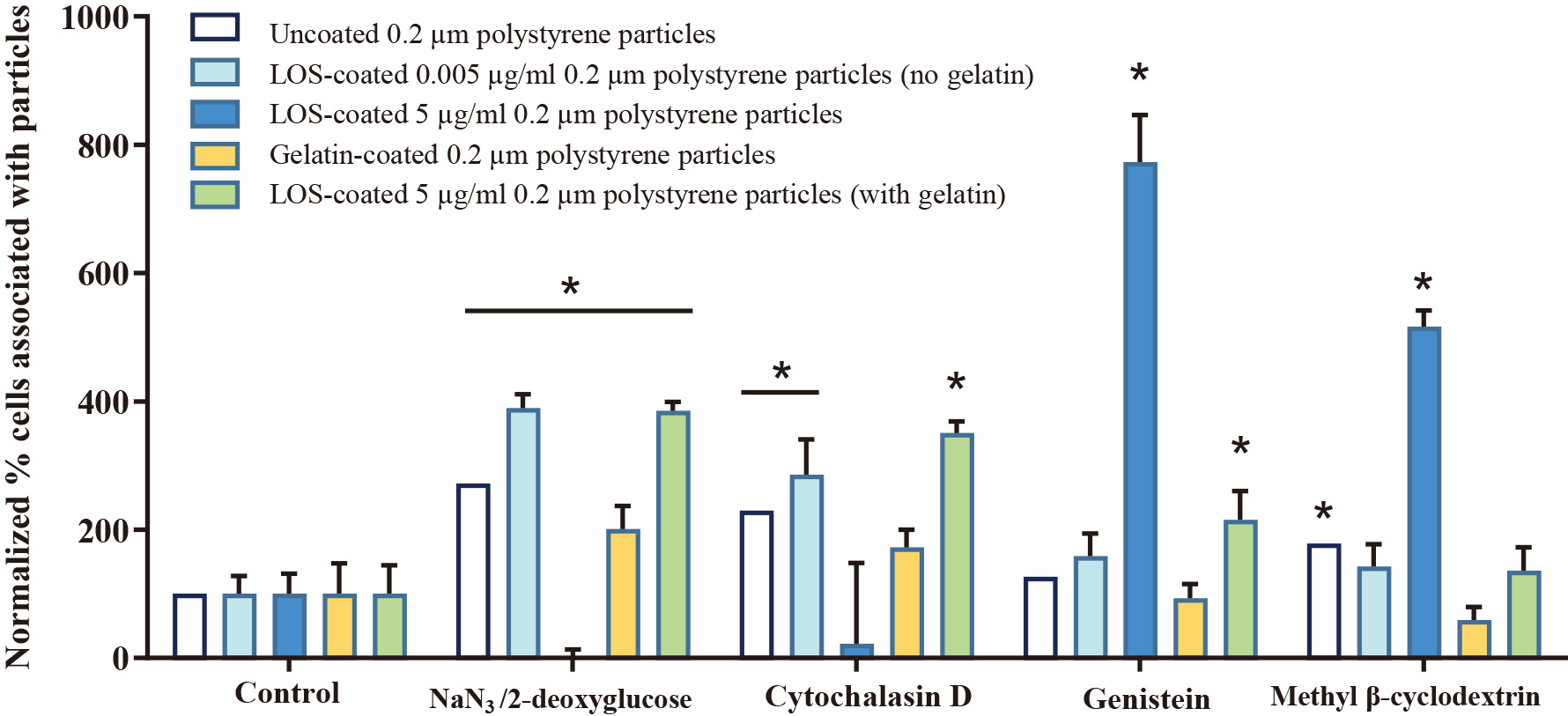
Further studies were conducted with specific pathway inhibitors to help elucidate specific uptake pathway for different particle types. Inhibitors of micropinocytosis (cytochalasin D) and caveolin-mediated endocytosis (genistein) did not inhibit the uptake of any particle type. Instead, particle uptake was sometimes enhanced with cell exposure to these inhibitors, suggesting that particles were taken up by multiple pathways. The inhibitor of cholesterol- dependent pathways (methyl β-cyclodextrin), which inhibits both caveolin-mediated and clathrin-mediated endocytosis, significantly reduced cell uptake of the 5 μg/mL LOS-coated particles. This suggests that the LOS-coated particles were primarily taken up by receptor-mediated endocytosis. However, due to the significant cell toxicity exhibited by the inhibitors of clathrin-mediated endocytosis, this could not be confirmed via chemical pathway inhibitors.
To prove that receptor-mediated endocytosis was the route by which LOS-coated particles were taken up into cells, we investigated whether PAFR-mediated endocytosis could be altered by the cognate ligand of the receptor (PAF). Upon cell incubation with PAF, the 5 μg/mL LOS-coated particles exhibited a 5- to 10-fold increase in uptake (Fig. 7). No significant change in the association of particles with lung cells was observed with the other particle types.
4. Discussion
Lipopolysaccharides (LPS), also known as endotoxins, are large lipid- and polysaccharide-containing molecules constituting the major component of the outer membrane of Gram-negative bacteria. They induce a strong immune response in animals and have been implicated in various pathogenic functions (Alexander and Rietschel, 2001; Simpson and Trent, 2019). Key studies have shown that isolating LPS from the bacterial surface and attaching it to a particle surface may not alter its functionality, enabling the particles to mimic some bacterial properties (Piazza et al., 2011). For example, titanium dioxide particles coated with LPS from E-coli induced pro-inflammatory signaling in primary human mononuclear phagocytes (Ashwood et al., 2007). LPS associated with polymeric and metallic components of orthopedic implants stimulate toll-like receptor (TLR) activation, which contributed to an inflammatory response (Greenfield et al., 2010; Hold and Bryant, 2011). However, the activation of the inflammatory response by LPS-coated particles was not desired for the current application.
Lipooligosaccharides (LOS) share a similar lipid A structure to LPS, but are lower in molecular weight, lack the O-antigens that allow serotyping, and have significantly lower toxicity profiles (Preston et al., 1996). LOS is the major glycolipid expressed on mucosal Gram-negative bacteria, including members of the genera Neisseria, Haemophilus, Bordetella, and Branhamella. It is a heat stable, amphipathic glycolipid complex that covers up to 75 % of the outer membrane (Silipo et al., 2011), and is crucial for the viability and survival of Gram-negative bacteria as it helps maintain proper outer membrane structure. It also contributes to host-bacterium interactions related to adherence, colonization, internalization, and ultimately survival in host cells.
The oligosaccharide region of LOS is highly heterogeneous within and between bacterial strains, containing both the recognition structures for host cells and components of the immune system, as well as carbohydrate moieties that may camouflage the bacterial surface from the host through molecular mimicry of host structures (Mandrell and Apicella, 1993; Masoud et al., 1997; Moran et al., 1996; Murphy and Apicella, 1987; Risberg et al., 1999a, 1999b; Silipo et al., 2011). In some bacterial species, these carbohydrate structures further facilitate bacterial adherence to the respiratory epithelium (Clark et al., 2012; Jalalvand and Riesbeck, 2014; Swords et al., 2000, 2001). Phosphorylcholine (ChoP) expressed as a terminal structure of the OS region facilitates adherence of multiple bacterial pathogens, including NTHi, to the platelet-activating factor receptor (PAFR) on bronchial cells by molecularly mimicking the structure of the cognate ligand platelet-activating factor (PAF) (Cundell et al., 1995; Gillespie et al., 1996; Harvey et al., 2001; Lysenko et al., 2000; Rosenow et al., 1997; Schweda et al., 2000; Weiser et al., 1997, 1998). As a potential targeting ligand for particles, our previous studies indicated that coupling ChoP-containing LOS from NTHi (strain 2019) to the surface of 1 μm particles facilitated significantly more binding to the human bronchial epithelial surface compared to particles coated with gelatin or coated with truncated LOS lacking the oligosaccharide chains that contain ChoP (Swords et al., 2000). Here, we aimed to evaluate the ability of ChoP-containing LOS to facilitate the uptake of smaller nanoparticles into respiratory epithelial cells.
Given the heterogeneity of the OS expression between bacterial strains, a screening of clinically isolated NTHi strains was performed to select a strain with high ChoP content to facilitate particle adherence and uptake into lung cells. Two of the three NTHi clinical isolates tested (956 and 3198) exhibited high ChoP activity. While the ChoP content of strain 956 had not been previously reported, the observed differences between strains 3198 and 2019 agreed with published literature, which has reported that 78 % of strain 3198 expressed ChoP with high reactivity, compared to 9 % of strain 2019 (Swords et al., 2000; Weiser et al., 1997). Based on these results and the known ability of NTHi 3198 to associate with and invade primary lung epithelial cells (Ketterer et al., 1999), LOS from NTHi 3198 was chosen as the ligand for further studies.
LOS is a large molecule containing a lipid structure that embeds in the bacterial outer membrane and an oligosaccharide structure that is exposed on the surface. The polystyrene particles used in this study exhibit a hydrophobic, negatively charged surface containing sulfate ester functional groups. Due to the hydrophobicity of the particles and the lipophilicity of the LOS lipid tails, we anticipated that the lipid tails of the LOS molecules would readily adsorb to the hydrophobic particle surface. In this case, the carbohydrate heads on the LOS structure containing ChoP could be available for cell targeting. Based on analysis of the zeta potential and XPS data, LOS was indeed coated onto the surface of polystyrene particles, particularly with the more highly concentrated LOS coating solution. However, the XPS data also suggested that the phosphate groups were buried within a mostly carbon-oxygen LOS backbone. This would be undesirable if the ChoP functional groups were not available to function as a targeting ligand on the particle surface. As XPS studies were conducted on dry particles, when the carbohydrate chains become hydrated, the LOS structure could open and give access to the ChoP moieties.
ITC further confirmed the ability of LOS to strongly adsorb to the nanoparticle surface. Typical enthalpies of binding for proteins to polystyrene nanoparticle surfaces has been reported to be on the order of 10 s to 100 s of kcal/mol (Porzeller et al., 2019), similar to the values obtained in this study. Using titration concentrations of 30 and 50 μg/mL, we estimated that there are about 34 LOS binding sites per particle. The titration concentration of 50 μg/mL (~7 μg/mL final concentration in the cell after titration), resulted in slightly more adsorbed LOS molecules than the 30 μg/mL titration concentration (~4 μg/mL final concentration in the cell after titration). This suggests that higher LOS concentrations enhance LOS adsorption to the particle surface. Previous work by Swords et al. reported a dose-response in which increasing the concentration of LOS coating resulted in more particle adherence to respiratory epithelium (Swords et al., 2000). LOS concentration is limited, though, as it readily forms micelles in aqueous solution at low concentrations due to its amphiphilic nature (Steimle et al., 2016). Thus, a higher LOS coating might be achieved by using a co-solvent to better solubilize the LOS. The ability of LOS to form micelles, as well as the complex structure of LOS, are potential reasons for the relatively high variability in the ITC data.
If the ChoP moieties on the LOS molecules remained accessible on the particle surface, we would expect the particles to exhibit better binding to and internalization into the respiratory epithelium. PAFR is expressed on human lung tissue membranes (Hwang et al., 1985; Shukla et al., 2016) and various lung epithelial cell lines (Chen et al., 2015). In the current study we confirmed PAFR expression in 16HBEo- bronchial epithelial cell monolayers via immuno-fluorescent staining and used these cells to evaluate the uptake of uncoated and coated particles. The 5 μg/mL LOS-coated particles exhibited the highest cell uptake of nanoparticles, with about 35 % uptake at the 24 h timepoint. All other particle types were taken up by less than 12 % of cells at any time. Confocal microscopy confirmed that particles were not just bound to the surface, but were internalized into the cell layers. While the ability of PAFR to mediate particle attachment to the respiratory epithelium had been previously established (Swords et al., 2000), this is the first time to our knowledge that bacterial-derived LOS has been used as a targeting ligand to facilitate particle uptake into cells.
We further investigated the uptake mechanism by blocking specific endocytic pathways using chemical inhibitors. NTHi bacteria have been shown to internalize into human lung epithelial cells by macropinocytosis and receptor-mediated endocytosis, so these pathways were likely candidates for LOS-coated nanoparticle uptake (Clementi and Murphy, 2011; Ketterer et al., 1999; Swords et al., 2000, 2001). Pre-incubation of the lung cells with NaN3-2-deoxyglucose completely knocked out their ability to take up the 5 μg/mL LOS-coated particles, suggesting an energy-dependent pathway for these particles, but had no impact on the other particle types. We further saw no evidence of the LOS-coated particles being taken up by macropinocytosis. Due to significant cell toxicity upon cell incubation with inhibitors of the clathrin-mediated pathway, we were unable to confirm this pathway using chemical inhibitors of endocytic pathways. Thus, a different approach was chosen in which the impact of a PAFR activator on the cellular uptake of uncoated and functionalized particles was investigated. A549 cells were pre-treated with PAF, the natural substrate for PAFR (Chao and Olson, 1993). Prior studies have shown that PAFR stimulation is involved in bacterial entry and could assist the delivery of siRNA to airway epithelia (Krishnamurthy et al., 2014; Zhang et al., 2000). In the current study, PAF pre-treatment significantly enhanced cell uptake of the 5 μg/mL LOS-coated particles, suggesting that PAF activation aided their uptake. Activation of PAFR via PAF exhibited no significant change on the association of any other particle type, demonstrating receptor targeting with the LOS coating.
In summary, the functionalization of polystyrene nanoparticles with LOS bacterial ligands resulted in better cellular uptake in PAFR-expressing lung cells. This strategy could be extended to drug-containing nanoparticles to enhance the therapeutic efficacy of drug compounds that require cellular internalization. Further, receptor-ligand interactions could be enhanced by activating the cell receptor, suggesting that enhanced cellular uptake could be achieved in disease states where the receptor is already activated, such as in the instance of infection, COPD, asthma, and cancer.
Acknowledgments
Funding for this work was provided by the National Institutes of Health (R21HL113876 and P30 DK054759). M. Timm was supported through an NSF-REU program in Nanoscience and Nanotechnology (NSF 1359063). We thank the University of Iowa Central Microscopy Research Facility, a core resource supported by the Vice President for Research & Economic Development, the Holden Comprehensive Cancer Center, and the Carver College of Medicine, for access to and training on the confocal and electron microscopes. Flow cytometry data presented were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa. The Facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. Part of the study has been published in the PhD thesis of author Mai Tu (Tu, 2015).
References
- Alexander C., Rietschel E.T., Bacterial lipopolysaccharides and innate immunity, Journal of endotoxin research, 7 (2001) 167–202. DOI: 10.1177/09680519010070030101
- Apicella M.A., Isolation and characterization of lipopolysaccharides, in: DeLeo F.R., Otto M. (Eds.), Bacterial Pathogenesis: Methods and Protocols, Humana Press, Totowa, NJ, 2008, pp. 3–13, ISBN: 978-1-60327-032-8. DOI: 10.1007/978-1-60327-032-8_1
- Ashwood P., Thompson R.P., Powell J.J., Fine particles that adsorb lipopolysaccharide via bridging calcium cations may mimic bacterial pathogenicity towards cells, Experimental Biology and Medicine, 232 (2007) 107–117. DOI: 10.3181/00379727-207-2320107
- Bagwell B., A journey through flow cytometric immunofluorescence analyses—Finding accurate and robust algorithms that estimate positive fraction distributions, Clinical Immunology Newsletter, 16 (1996) 33–37. DOI: 10.1016/S0197-1859(00)80002-3
- Campagnari A.A., Gupta M.R., Dudas K.C., Murphy T.F., Apicella M.A., Antigenic diversity of lipooligosaccharides of nontypable Haemophilus influenzae, Infection and Immunity, 55 (1987) 882–887. DOI: 10.1128/iai.55.4.882-887.1987
- Chao W., Olson M.S., Platelet-activating factor: receptors and signal transduction, Biochemical Journal, 292 (1993) 617–629. DOI: 10.1042/bj2920617
- Chen J., Lan T., Zhang W., Dong L., Kang N., Zhang S., Fu M., Liu B., Liu K., Zhan Q., Feed-forward reciprocal activation of PAFR and STAT3 regulates epithelial–mesenchymal transition in non–small cell lung cancer, Cancer Research, 75 (2015) 4198–4210. DOI: 10.1158/0008-5472.CAN-15-1062
- Clark S.E., Snow J., Li J., Zola T.A., Weiser J.N., Phosphorylcholine allows for evasion of bactericidal antibody by Haemophilus influenzae, PLoS Pathogens, 8 (2012) e1002521. DOI: 10.1371/journal.ppat.1002521
- Clementi C., Murphy T., Non-typeable Haemophilus influenzae invasion and persistence in the human respiratory tract, Frontiers in Cellular and Infection Microbiology, 1 (2011) DOI: 10.3389/fcimb.2011.00001
- Cochi S.L., Broome C.V., Vaccine prevention of Haemophilus influenzae type b disease: past, present and future, Pediatric Infectious Disease, 5 (1986) 12–19. DOI: 10.1097/00006454-198601000-00003
- Cundell D.R., Gerard N.P., Gerard C., Idanpaan-Heikkila I., Tuomanen E.I., Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor, Nature, 377 (1995) 435–438. DOI: 10.1038/377435a0
- Fenton O.S., Olafson K.N., Pillai P.S., Mitchell M.J., Langer R., Advances in biomaterials for drug delivery, Advanced Materials, 30 (2018) 1705328. DOI: 10.1002/adma.201705328
- Foxwell A.R., Kyd J.M., Cripps A.W., Nontypeable Haemophilus influenzae: pathogenesis and prevention, Microbiology and Molecular Biology Reviews, 62 (1998) 294–308. DOI: 10.1128/MMBR.62.2.294-308.1998
- Galanos C., Lüderitz O., Westphal O., A new method for the extraction of R lipopolysaccharides, European Journal of Biochemistry, 9 (1969) 245–249. DOI: 10.1111/j.1432-1033.1969.tb00601.x
- Gillespie S., Ainscough S., Dickens A., Lewin J., Phosphorylcholine-containing antigens in bacteria from the mouth and respiratory tract, Journal of Medical Microbiology, 44 (1996) 35–40. DOI: 10.1099/00222615-44-1-35
- Greenfield E.M., Beidelschies M.A., Tatro J.M., Goldberg V.M., Hise A.G., Bacterial pathogen-associated molecular patterns stimulate biological activity of orthopaedic wear particles by activating cognate Toll-like receptors, Journal of Biological Chemistry, 285 (2010) 32378–32384. DOI: 10.1074/jbc.M110.136895
- Gruenert D.C., Basbaum C.B., Welsh M.J., Li M., Finkbeiner W.E., Nadel J.A., Characterization of human tracheal epithelial cells transformed by an origin-defective simian virus 40, proceedings of the National Academy of Sciences of the United States of America, 85 (1988) 5951–5955. DOI: 10.1073/pnas.85.16.5951
- Harvey H.A., Swords W.E., Apicella M.A., The Mimicry of human glycolipids and glycosphingolipids by the lipooligosaccharides of pathogenic Neisseria and Haemophilus, Journal of Autoimmunity, 16 (2001) 257–262. DOI: 10.1006/jaut.2000.0477
- Hold G.L., Bryant C.E., The molecular basis of lipid a and Toll-like receptor 4 interactions, in: Knirel Y.A., Valvano M.A. (Eds.), Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Interaction with Host Cells, Springer Vienna, Vienna, 2011, pp. 371–387, ISBN: 978-3-7091-0733-1. DOI: 10.1007/978-3-7091-0733-1_12
- Hwang S.-B., Lam M.-H., Shen T., Specific binding sites for platelet activating factor in human lung tissues, Biochemical and Biophysical Research Communications, 128 (1985) 972–979. DOI: 10.1016/0006-291x(85)90142-1
- Jalalvand F., Riesbeck K., Haemophilus influenzae: recent advances in the understanding of molecular pathogenesis and polymicrobial infections, Current Opinion in Infectious Diseases, 27 (2014) 268–274. DOI: 10.1097/qco.0000000000000057
- Ketterer M.R., Shao J.Q., Hornick D.B., Buscher B., Bandi V.K., Apicella M.A., Infection of primary human bronchial epithelial cells by Haemophilus influenzae: Macropinocytosis as a mechanism of airway epithelial cell entry, Infection and Immunity, 67 (1999) 4161–4170. DOI: 10.1128/IAI.67.8.4161-4170.1999
- Krishnamurthy S., Behike M.A., Apicella M.A., McCray P.B., Davidson B.L., Platelet activating factor receptor activation improves siRNA uptake and RNAi responses in well-differentiated airway epithelia, Molec Ther - Nucleic Acids, 3 (2014) e175. DOI: 10.1038/mtna.2014.26
- Lysenko E., Richards J.C., Cox A.D., Stewart A., Martin A., Kapoor M., Weiser J.N., The position of phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae affects binding and sensitivity to C-reactive protein-mediated killing, Molecular Microbiology, 35 (2000) 234–245. DOI: 10.1046/j.1365-2958.2000.01707.x
- Mandrell R.E., Apicella M.A., Lipo-oligosaccharides (LOS) of mucosal pathogens: molecular mimicry and host-modification of LOS, Immunobiology, 187 (1993) 382–402. DOI: 10.1016/s0171-2985(11)80352-9
- Masoud H., Moxon E.R., Martin A., Krajcarski D., Richards J.C., Structure of the variable and conserved lipopolysaccharide oligosaccharide epitopes expressed by Haemophilus influenzae serotype b strain Eagan, Biochemistry, 36 (1997) 2091–2103. DOI: 10.1021/bi961989y
- Moran A.P., Prendergast M.M., Appelmelk B.J., Molecular mimicry of host structures by bacterial lipopolysaccharides and its contribution to disease, FEMS Immunology and Medical Microbiology, 16 (1996) 105–115. DOI: 10.1111/j.1574-695X.1996.tb00127.x
- Murphy T.F., Apicella M.A., Nontypable Haemophilus influenzae: a review of clinical aspects, surface antigens, and the human immune response to infection, Reviews of Infectious Diseases, 9 (1987) 1–15. DOI: 10.1093/clinids/9.1.1
- Pang B., Winn D., Johnson R., Hong W., West-Barnette S., Kock N., Swords W.E., Lipooligosaccharides containing phosphorylcholine delay pulmonary clearance of nontypeable Haemophilus influenzae, Infection and Immunity, 76 (2008) 2037–2043. DOI: 10.1128/IAI.01716-07
- Patra J.K., Das G., Fraceto L.F., Campos E.V.R., Rodriguez-Torres M.d.P., Acosta-Torres L.S., Diaz-Torres L.A., Grillo R., Swamy M.K., Sharma S., Habtemariam S., Shin H.-S., Nano based drug delivery systems: recent developments and future prospects, Journal of Nanobiotechnology, 16 (2018) 71. DOI: 10.1186/s12951-018-0392-8
- Piazza M., Colombo M., Zanoni I., Granucci F., Tortora P., Weiss J., Gioannini T., Prosperi D., Peri F., Uniform lipopolysaccharide (LPS)-loaded magnetic nanoparticles for the investigation of LPS-TLR4 signaling, Angewandte Chemie International Edition, 50 (2011) 622–626. DOI: 10.1002/anie.201004655
- Porzeller D., Morsbach S., Landfester K., Isothermal titration calorimetry as a complementary method for investigating nanoparticle-protein interactions, Nanoscale, 11 (2019) 19265–19273. DOI: 10.1039/C9NR05790K
- Preston A., Mandrell R.E., Gibson B.W., Apicella M.A., The lipooligosaccharides of pathogenic gram-negative bacteria, Critical Reviews in Microbiology, 22 (1996) 139–180. DOI: 10.3109/10408419609106458
- Risberg A., Alvelius G., Schweda E.K., Structural analysis of the lipopolysaccharide oligosaccharide epitopes expressed by Haemophilus influenzae strain RM. 118-126, European Journal of Biochemistry, 265 (1999a) 1067–1074. DOI: 10.1046/j.1432-1327.1999.00832.x
- Risberg A., Masoud H., Martin A., Richards J.C., Moxon E.R., Schweda E.K., Structural analysis of the lipopolysaccharide oligosaccharide epitopes expressed by a capsule-deficient strain of Haemophilus influenzae Rd, European Journal of Biochemistry, 261 (1999b) 171–180. DOI: 10.1046/j.1432-1327.1999.00248.x
- Roederer M., Treister A., Moore W., Herzenberg L.A., Probability binning comparison: a metric for quantitating univariate distribution differences, Cytometry, 45 (2001) 37–46. DOI: 10.1002/1097-0320(20010901)45:1<37::aid-cyto1142>3.0.co;2-e
- Rosenow C., Ryan P., Weiser J.N., Johnson S., Fontan P., Ortqvist A., Masure H.R., Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae, Molecular Microbiology, 25 (1997) 819–829. DOI: 10.1111/j.1365-2958.1997.mmi494.x
- Schweda E.K., Brisson J.R., Alvelius G., Martin A., Weiser J.N., Hood D.W., Moxon E.R., Richards J.C., Characterization of the phosphocholine-substituted oligosaccharide in lipopolysaccharides of type b Haemophilus influenzae, European Journal of Biochemistry, 267 (2000) 3902–3913. DOI: 10.1046/j.1432-1327.2000.01426.x
- Shukla S.D., Muller H.K., Latham R., Sohal S.S., Walters E.H., Platelet-activating factor receptor (PAFr) is upregulated in small airways of alveoli of smokers and COPD patients, Respirology, 21 (2016) 504–510. DOI: 10.1111/resp.12709
- Silipo A., Molinaro A., Lipid a structure, in: Knirel Y.A., Valvano M.A. (Eds.), Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Interaction with Host Cells, Springer Vienna, Vienna, 2011, pp. 1–20, ISBN: 978-3-7091-0733-1. DOI: 10.1007/978-3-7091-0733-1_1
- Simpson B.W., Trent M.S., Pushing the envelope: LPS modifications and their consequences, Nature Reviews Microbiology, 17 (2019) 403–416. DOI: 10.1038/s41579-019-0201-x
- St. Geme J.W., Molecular determinants of the interaction between Haemophilus influenzae and human cells, American Journal of Respiratory and Critical Care Medicine, 154 (1996) S192–S196. DOI: 10.1164/ajrccm/154.4_Pt_2.S192
- Starner T.D., Zhang N., Kim G., Apicella M.A., Paul B., McCray J., Haemophilus influenzae forms biofilms on airway epithelia: implications in cystic fibrosis, American Journal of Respiratory and Critical Care Medicine, 174 (2006) 213–220. DOI: 10.1164/rccm.200509-1459OC
- Steimle A., Autenrieth I.B., Frick J.-S., Structure and function: lipid A modifications in commensals and pathogens, International Journal of Medical Microbiology, 306 (2016) 290–301. DOI: 10.1016/j.ijmm.2016.03.001
- Swords W.E., Buscher B.A., Ver Steeg II K., Preston A., Nichols W.A., Weiser J.N., Gibson B.W., Apicella M.A., Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor, Molecular Microbiology, 37 (2000) 13–27. DOI: 10.1046/j.1365-2958.2000.01952.x
- Swords W.E., Ketterer M.R., Shao J., Campbell C.A., Weiser J.N., Apicella M.A., Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling, Cellular Microbiology, 3 (2001) 525–536. DOI: 10.1046/j.1462-5822.2001.00132.x
- Tu M., Lipooligosaccharide-modified polymeric particles for targeted pulmonary drug delivery, Ph.D., University of Iowa, 2015. DOI: 10.17077/etd.21vj0y3n
- Turk D.C., The pathogenicity of Haemophilus influenzae, Journal of Medical Microbiology, 18 (1984) 1–16. DOI: 10.1099/00222615-18-1-1
- Weiser J.N., Goldberg J.B., Pan N., Wilson L., Virji M., The phosphorylcholine epitope undergoes phase variation on a 43-kilodalton protein in Pseudomonas aeruginosa and on pili of Neisseria meningitidis and Neisseria gonorrhoeae, Infection and Immunity, 66 (1998) 4263–4267. DOI: 10.1128/iai.66.9.4263-4267.1998
- Weiser J.N., Shchepetov M., Chong S.T., Decoration of lipopolysaccharide with phosphorylcholine: a phase-variable characteristic of Haemophilus influenzae, Infection and Immunity, 65 (1997) 943–950. DOI: 10.1128/iai.65.3.943-950.1997
- Zhang J.-R., Mostov K.E., Lamm M.E., Nanno M., Shimida S.-i., Ohwaki M., Tuomanen E., The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells, Cell, 102 (2000) 827–837. DOI: 10.1016/S0092-8674(00)00071-4
Nomenclature
BHI
Brain heart infusion
BSA
Bovine serum albumin
ChoP
Phosphorylcholine
ELISA
Enzyme-linked immunosorbent assay
ITC
Isothermal titration calorimetry
LOS
Lipooligosaccharide
LPS
Lipopolysaccharide
NAD
Nicotinamide adenine dinucleotide
NTHi
Non-typeable Haemophilus influenzae
PAF
Platelet-activating factor
PAFR
Platelet-activating factor receptor
pNPP
Nitrophenyl phosphate bis (cyclohexyl ammonium)
SDS
Sodium dodecyl sulfate
SEM
Scanning electron microscope
XPS
X-ray photoelectron spectrometer
Authors’ Short Biographies
Mai H. Tu

Mai H. Tu earned a Ph.D. in pharmaceutics from the University of Iowa, USA under the guidance of Dr. Jennifer Fiegel. Her doctoral work focused on developing lipooligosaccharide-modified polymeric particles for targeted pulmonary drug delivery.
Timothy M. Brenza

Timothy M. Brenza completed a postdoctoral fellowship under the guidance of Dr. Jennifer Fiegel from 2008–2010. He is currently an Assistant Professor of chemical and biological engineering at South Dakota School of Mines and Technology.
Margaret R. Ketterer

Margaret R. Ketterer has worked at the University of Iowa Department of Microbiology and Immunology, in the laboratory of Dr. Michael Apicella, since 1993. Her research efforts have concentrated on the role of bacterial lipo-oligosaccharides in infections of human cells by Neisseria species and by Haemophilus influenzae.
Morgan Timm

Morgan Timm received a B.S. degree in biochemistry and molecular biology from Gustavus Adolphus College, USA and is currently an MD/PhD student at Washington University, USA. She conducted a summer research project in the laboratory of Dr. Jennifer Fiegel as part of the Nano NSF REU program at the University of Iowa.
Benjamin M. King

Benjamin M. King earned a Ph.D. in chemical and biochemical engineering from the University of Iowa, USA in 2018 under the guidance of Dr. Jennifer Fiegel. His doctoral work focused on interactions of environmental and therapeutics particles with the airway microenvironment.
Michael A. Apicella

Michael A. Apicella is a Professor Emeritus of microbiology and immunology at the University of Iowa. His research focused on understanding the factors involved in human infections caused by Gram-negative bacterial pathogens.
Jennifer Fiegel

Jennifer Fiegel is a Professor of chemical and biochemical engineering at the University of Iowa (Iowa City, Iowa), with a secondary appointment in pharmaceutical sciences and experimental therapeutics. Her research expertise is in the design of improved therapeutic drug delivery strategies for the treatment and prevention of infections.
 https://ror.org/036jqmy94
https://ror.org/036jqmy94 


;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_08.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_09.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_10.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_11.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_12.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_13.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/41_2024005_14.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)