Tunability of Mg2Si Bandgap by Formation of Mg2(Si, C) with an Anti-Fluorite Structure Examined by First-Principles Calculations
2019 Volume 60 Issue 9 Pages 1873-1880

Details
2019 Volume 60 Issue 9 Pages 1873-1880
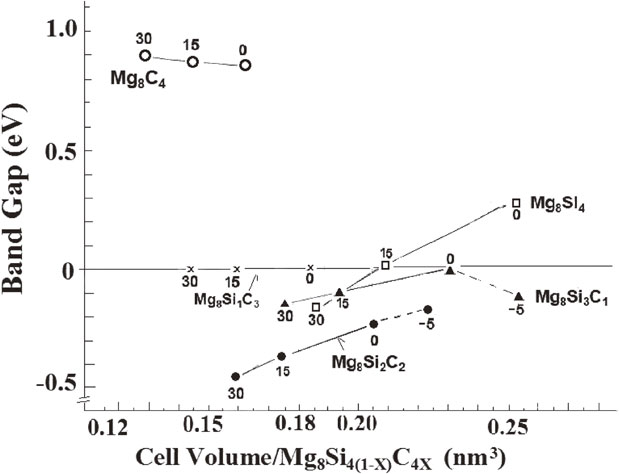
We used first-principles calculations to investigate the effects of replacing Si atoms in Mg2Si with C atoms to tune its bandgap and enhance its thermoelectric performance. First-principles calculations suggest that the substitution of Si by C atoms in the Mg2Si lattice forms Mg8Si4−zCz (z = 1, 2, and 3) with a sustained anti-fluorite structure, which results in an unexpected bandgap contraction. However, the bandgap of Mg8Si4(1−x)C4x composed of a supercell structure of a rhombohedral Mg2Si primitive cell and Si-substitution with C has a wide bandgap when the C/Si ratio is sufficiently high. The formation enthalpies from Mg, Si, and C (diamond) are negative under pressures greater than ca. 15 GPa.