1. Introduction
Manufacturing of nonferrous metals, e.g., copper (Cu) and zinc (Zn), produces hazardous by-products such as arsenic (As) and cadmium (Cd) because their ores and concentrates contain those hazardous elements. For example, Cu concentrate commonly contains several hundreds to tens of hundreds mass ppm of As, and the global amount of As in the Cu concentrate supplied for Cu smelting in a year is as high as 150,000 tons.1) The worldwide circulation of As, principally as arsenic trioxide (As2O3), is 25,000 tons in a year in As-equivalent.2) Metallic Cd is a by-product of Zn production and 25,000 tons are manufactured yearly.2)
The recent lowering of ore grades has involved increase of concentrations of impurity As and Cd in concentrates; therefore, future production of those hazardous by-products is expected to increase. In the past, As compounds were widely used in pesticides, herbicides, fungicides, and wood preservers,3) as well as in fining agents for accelerated bubble removal from glass.4) Cd compounds were formerly used in Ni–Cd batteries, pigments, and high-corrosion-resistant plating.5) However, usage regulations, such as RoHS directive and REACH regulations in EU region, emphasizing human safety and reduced environmental impact are expected to decrease consumption of these hazardous by-products. The reduction of hazardous by-product consumption concurrent with their increasing production is a significant problem for the nonferrous manufacturing industry. Thus, novel uses of these hazardous by-products must be explored to secure a continuous and non-problematic supply of nonferrous metals.
One possibility is to use hazardous elements as components of materials with outstanding functionality in strictly and safely controlled facilities. In particular, materials related to renewable-energy power generation systems are likely acceptable to the society, considering the balance between their risks and benefits. One such example is the CdTe thin-film solar cells used in solar power stations. In 2019, it accounted for 77% of the thin-film solar cells manufactured globally (in detail, CdTe: 5.7 GWp, Cu(In,Ga)Se2: 1.6 GWp, and amorphous Si: 0.2 GWp),6) although it contains toxic Cd. Furthermore, lead halide perovskites, which enable easy fabrication of solar cells exhibiting high conversion efficiency of more than 23%7–9) are currently under development. In the case of As, although GaAs thin-film solar cells exhibit the best conversion efficiency among single-junction solar cells (29.1%),10) their manufacturing scale is relatively small. Therefore, it is necessary to find an appropriate use for As as an energy material.
Enargite, i.e., Cu3AsS4, is a I3–V–VI4 compound semiconductor possessing a wurtzite-related structure11,12) with CuS4 and AsS4 tetrahedra in a vertex-sharing linkage. Further, enargite is one of the minerals found in Cu ore; therefore, As elimination from enargite (as well as tennantite, i.e., Cu12As4S13) through roasting and acid treatment has been studied.13,14) Enargite has a history of causing As contamination of rivers due to Cu mining development.15) However, it has been studied as a solar-cell absorber since 1995, when Pauporte et al. reported its appropriate band gap to achieve maximum conversion efficiency of a single-junction solar cell according to Shockley–Queisser theory,16) and its exhibition of p-type electrical conduction with conductivity of 0.1 S cm−1 at room temperature.17) Calculation of its electronic structure,18–20) electrical and optical properties,21,22) and thin-film fabrication23) have recently been reported, and life cycle assessment of Cu3AsS4 solar cells has been performed.24) Thus, research on Cu3AsS4 as a solar-cell absorber has been progressing steadily.
In general, p-n homojunction solar cells have an advantage to achieve high conversion efficiency compared to heterojunction cells, as they are free from unfavorable energy-band discontinuity and favor ideal interface formation with fewer interfacial defects. In the case of Cu3AsS4, high n-type electrical conductivity is not expected. This is because the conduction band minimum (CBM) of this material, which mainly consists of the antibonding state of S 3p and As 4s, exhibits quite small dispersion, thereby indicating a large electron effective mass.20) According to a first principles calculation by Ma et al.,20) the Bader charges of the constituent atoms are +0.5, +1.09, and −0.61 for Cu, As, and S, respectively, suggesting high covalency of the As–S bonding. Therefore, it appears that Cu3AsS4 is not a complex sulfide of Cu+ and As5+, but rather a copper(I) thioarsenate consisting of Cu+ and AsS43−. This may explain the large contribution of the S 3p states to the CBM. Note that this characteristic differs from those of common chalcogenide compound semiconductors such as ZnS, of which the CBM mainly consists of Zn 4s states.25–27)
Compared to As–S bonding, however, As–O bonding should exhibit a highly ionic nature as electronegativity of O is higher than that of S. Thus, if enargite-type Cu3AsO4 were obtained, its CBM would likely consist of Cu 4s and/or As 4s states and exhibit large dispersion, thereby resulting in a suitable electronic structure for n-type electrical conduction. However, to the best of our knowledge, enargite-type Cu3AsO4 has yet to be obtained.28)
In this study, the electronic structure of enargite-type Cu3AsO4 is theoretically studied through first principles calculations using the generalized gradient approximation (GGA) functional with corrections for the on-site Coulomb interactions (GGA+U). We note that introduction of corrections for on-site Coulomb interactions is a method of correcting the strong electron-correlation of Cu 3d electrons and does not require high computational cost. In this work, convergence is achieved for geometry optimization of the enargite-type Cu3AsO4, indicating that this material is evaluated as practically stable in the calculation. The thermodynamical stability of enargite-type Cu3AsO4 is evaluated through comparison of the calculated formation enthalpy for 17 compounds appearing in the Cu–As–O ternary system. The Cu 4s and Cu 3d states are shown to strongly contribute to the CBM and valence band maximum (VBM), respectively, similar to other oxide semiconductors containing Cu+. The effective masses of electron (me*/m0) and hole (mh*/m0) in enargite-type Cu3AsO4 are 0.50 and 2.5, respectively, indicating that both n-type and p-type electrical conduction are expected by appropriate impurity doping in contrast to Cu3AsS4 with presumed lack of high n-type electrical conduction. The calculated optical absorption coefficient of Cu3AsO4 just above the band gap is of the order of 104 cm−1. These results indicate that Cu3AsO4 is promising as a solar-cell absorber material enabling p–n homojunction. Finally, synthesis of enargite-type Cu3AsO4 from Cu2O and As2O5 by the mechanical alloying method is examined. Although the solid-state reaction does not yield Cu3AsO4 formation, the mechanical alloying produces a small amount of stannite-type Cu3AsO4, which is one of the polymorphs of this I3–V–VI4 semiconductor, creating an expectation that single-phase enargite-type Cu3AsO4 can be synthesized.
2. Methods
2.1 Computational method
DFT calculations were performed with a plane-wave-based pseudo-potential method implemented in the CASTEP code.29) All DFT calculations were performed using the norm-conserving pseudopotentials generated by the CASTEP code, and the Perdew–Burke–Ernzerhof for solids (PBEsol),30) one of the GGA functional optimized for solid state materials and surface, was adopted for exchange and correlation functional. The Monkhorst-Pack-grid31,32) were applied to brillouin-zone sampling and the mesh grid was set to 4 × 5 × 5, 6 × 6 × 8, 4 × 4 × 4, and 3 × 4 × 4 k-point meshes for enargite-type, stannite-type, and Ag3PO4-type Cu3AsO4, and enargite-type Cu3AsS4, respectively. The plane-wave cutoff energy was set to 1500 eV for all calculations. On self-consistent calculations, the ensemble density-functional theory33,34) was used for electronic minimizer in order to achieve efficient convergence. Geometry optimization was performed with Broyden–Fletcher–Goldfarb–Shanno algorism35) with the imposed symmetry based on the corresponding space group. In order to estimate formation enthalpy of assumed compounds, not only atomic positions but also lattice parameters were relaxed. The convergence conditions of geometry optimization were as follows: energy convergence tolerance: 5.0 × 10−5 eV atom−1, maximum ionic displacement tolerance: 5.0 × 10−3 Å, maximum force tolerance: 1.0 × 10−2 eV Å−1, and maximum stress tolerance: 2.0 × 10−1 GPa.
The Hubbard correction for on-site Coulomb interactions (GGA+U)36) was introduced for the Cu 3d electrons in calculations of detailed electronic structures of the enargite-type Cu3AsO4 and Cu3AsS4. Note that stability of each crystalline phases was considered with consideration of U. The value of U was set to 4 eV as a U of 4–6 eV is typically employed in GGA+U calculations for Cu+-containing oxides and sulfides.37–40)
The initial structures subjected to geometry optimization for enargite-type, stannite-type, and Ag3PO4-type Cu3AsO4 were based on the lattice parameters and atomic coordination of the corresponding isostructural oxides: enargite-type Li3AsO4 (ICSD#75927), stannite-type Cu3VO4 (ICSD#418372), and Ag3PO4-type Ag3AsO4 (ICSD#76969), respectively. To obtain the formation enthalpies of compounds, calculations on simple substances (Cu, As, and O2) were performed for the face-centered cubic Cu, trigonal As, and isolated O2 molecule.
2.2 Mechanical alloying
Starting reagents of Cu2O (99.5%; Fujifilm Wako Pure Chemical Corporation, Japan) and As2O5 (99.9%; Strem Chemicals, US) were weighed at a molar ratio of 3:1. Then, 3 g of mixed powder and 78 g of zirconia balls with 10-mm diameter were placed in a zirconia pod with 45-mL capacity, which was then mounted in a stainless-steel gas-tight jacket. The inner space of the jacket was evacuated via rotary pump and then filled with Ar gas at 5 atm. The pods were rotated at 700 rpm in a planetary ball mill (Pulverisette-7, Fritsch, Germany). Following 30 min of ball milling, approximately 10 mg of the powder was sampled for analysis; the remainder was subjected to another round of continuous ball milling following evacuation and re-filling of the jacket with Ar gas, and this process was repeated for 4 times in total. The phases in the obtained powder were identified by powder X-ray diffraction (XRD, SmartLab, Rigaku, Japan).
3. Results and Discussion
3.1 Stability of enargite-type Cu3AsO4 among plausible polymorphs
Cu3AsO4, the synthesis of which has not been reported to date,28) possibly possesses the following three crystal structures based on comparison with the analogue compounds. The first is the enargite-type structure shown in Fig. 1(a), which consists of CuO4 and AsO4 tetrahedra with a vertex-sharing linkage. This is one of the ternary structures derived from the wurtzite-type structure, in which the O is arranged in hexagonal closest packing. As well as Cu3AsS4, both Li3AsO4 and Na3AsO4 have this structure. The second is the stannite-type structure shown in Fig. 1(b), which again consists of CuO4 and AsO4 tetrahedra. However, this is one of the ternary structures derived from the binary zincblende-type structure, in which the O is arranged in cubic closest packing. Ag3VO4 and Cu3VO4 also possess this stannite-type structure. The final structure is the Ag3PO4-type shown in Fig. 1(c), which consists of AsO4 tetrahedra and almost planar-coordinate AgO4 (CuO4 in the case of Cu3AsO4). In addition to Ag3PO4, Ag3AsO4 possesses this structure.

Convergence was achieved for geometry optimization of Cu3AsO4 with these three polymorphic structures using GGA (without U) in first principles calculations. This result indicates that these polymorphs all can exist stably on a computer. The calculated lattice parameters and atomic coordinates are listed in Tables A1–A4 in the Appendix. The formation enthalpies of these three polymorphs according to reaction (1) below are reported in Table 1.
| \begin{equation}
\text{3Cu} + \text{As} + \text{2O$_{2}$} \to \text{Cu$_{3}$AsO$_{4}$}
\end{equation}
| (1) |

Table 1 Cu3AsO4 formation enthalpies for polymorphic structures calculated by GGA without U.
The lowest formation enthalpy was obtained for the enargite-type structure and, thus, this structure is expected to be the most stable; however, only small differences in formation enthalpy were noted, at only 17 and 31 kJ mol−1 between the enargite-type and stannite-type and between the enargite-type and Ag3PO4-type polymorphs, respectively. Considering the existence of β-CuGaO2, the formation enthalpy of which is ∼40 kJ mol−1 higher than that of stable-phase α-CuGaO2,37,41) this result does not exclude the possibility of the formation of stannite-type and Ag3PO4-type Cu3AsO4.
The calculated lattice parameters of the enargite-type Cu3AsO4 with and without U are approximately identical (Table 2). These values are smaller than the experimentally determined values for Na3AsO4 and Cu3AsS4, and larger than those for Li3AsO4. This trend is reasonable considering the corresponding ionic radii (Cu+: 0.60 Å, Li+: 0.59 Å, Na+: 0.99 Å, O2− 1.40 Å, S2− 1.84 Å);42) thus, the obtained structure seems to be appropriate.
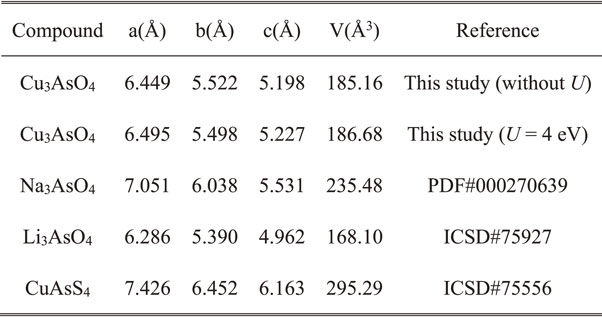
Table 2 Lattice parameters of enargite-type oxides and sulfide.
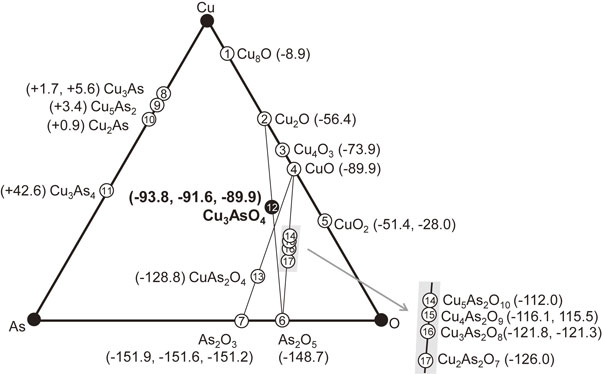
Figure 2 shows the ternary phase diagram of the Cu–As–O system, in which the materials with the calculated formation enthalpies at 0 K are presented. Multiple values are provided for the polymorphs. As the number of atoms in the chemical formula varies for each substance, the formation enthalpies are normalized by the number of atoms (i.e., kJ mol-of-atom−1). The results are also summarized in Table A6 in the Appendix, which includes the initial structural data subjected to geometry optimization. The formation enthalpies calculated for the oxides are all negative. That of the enargite-type Cu3AsO4 is −93.8 kJ mol-of-atom−1, which is slightly larger (i.e., this material is thermodynamically less stable) than those of the CuO–As2O5 and CuO–As2O3 compounds with similar composition (e.g., Cu3As2O8 and CuAs2O4 etc.). Judging from the three-phase tie-triangles, the enargite-type Cu3AsO4 is a metastable phase at 0 K considering that convergence is achieved for its geometry optimization at 0 K as described above. The high-temperature solid-state reaction at 600–1000 °C between Cu2O and As2O5 did not yield formation of Cu3AsO4, but Cu2O, Cu3As2O8, and As2O3 were formed (see Fig. A1 of Appendix). This result also implies that the enargite-type Cu3AsO4 is not a thermodynamically stable at 600–1000 °C. However, considering the fact that the difference in the calculated formation enthalpies at 0 K between Cu3AsO4 and other compounds with similar composition is not large, at approximately 20–30 kJ mol-of-atom−1, it is implied that enargite-type Cu3AsO4 can be synthesized as a metastable phase. The synthesis method of Cu3AsO4 is again discussed below, in Section 3.5, in the context of a mechanical alloying trial.
Figures 3(a) and (b) show the electronic band structure along the symmetry line and the atomically projected partial density of states (DOS) of enargite-type Cu3AsO4, respectively, as calculated via GGA+U with U = 4 eV. The CBM and VBM are located at the Γ- and X-points, respectively, indicating that enargite-type Cu3AsO4 is an indirect semiconductor; this is in contrast to enargite-type Cu3AsS4 with a direct band gap.20,43) As the energy at the top of the valence band at the Γ-point is only −0.30 eV below that of the VBM, the difference between the minimum indirect band gap and direct gap of Cu3AsO4 is very small.

The region just above the CBM of enargite-type Cu3AsO4 is mainly composed of Cu 4s states and exhibits a large dispersion. This is typical for oxide semiconductors consisting of cations with d10s0 electronic configuration, in which the spherically symmetric s-state of the cations contributes strongly to the region around the CBM and causes large dispersion.44) The effective electron mass (me*) calculated from the parabolic approximation of the conduction band edge was 0.50 m0 (Table 3). This value is sufficiently small, similar to those of typical oxide semiconductors consisting of cations with d10s0 electronic configurations such as ZnO (0.28 m0)45) and SnO2 (0.4 m0).46)

Table 3 Effective masses (in units of free electron mass, m0) of electrons and holes for enargite-type Cu3AsO4 and Cu3AsS4.
The region just below the VBM of enargite-type Cu3AsO4 is dominantly composed of Cu 3d states. This is due to the high energy of the Cu 3d states and is a common feature of Cu+-containing oxide and sulfide semiconductors such as Cu2O, α-CuAlO2, and CuInS2.47–49) The effective hole mass (mh*) calculated from the valence band edge is 2.5 m0. For example, α-CuAlO2, which is a p-type oxide semiconductor, has a similar effective hole mass (mh*/m0 = 2.5) and exhibits an electronic conductivity of ∼0.5 S cm−1 with a carrier concentration of 5–10 × 1017 cm−3.50–52) Therefore, enargite-type Cu3AsO4 is expected to exhibit enough p-type conductivity as an electrode or solar cell, although its mh* is indeed larger than those of typical III–V compound semiconductors. Thus, enargite-type Cu3AsO4 is expected to exhibit both n- and p-type conductions (i.e., bipolar conduction) by appropriate doping, thereby enabling a p-n homojunction.
For comparison, the electronic structure of enargite-type Cu3AsS4 was calculated by GGA+U with U = 4 eV, as shown in Figs. 3(c) and (d). The obtained lattice parameters are in good agreement with the experimentally determined values, with a difference of less than 2% (see Table A5 in the Appendix). The Cu3AsS4 conduction band possesses a distinctly different electronic structure from that of Cu3AsO4. The region just above the CBM of Cu3AsS4 is mainly composed of the antibonding state of S 3p and As 4s and exhibits a very small dispersion similar to the previous reports.20,43) This is in contrast to that of Cu3AsO4, which exhibits large dispersion. As noted in the introduction, Cu3AsS4 possesses Bader charges of +0.5 for Cu, +1.09 for As, and −0.61 for S; therefore, this material does not appear to be a complex sulfide of Cu+ and As5+, but rather a copper (I) thioarsenate consisting of Cu+ and AsS43−. This trend is also apparent from the Mulliken charges listed in Table 4, although these values are slightly smaller than the corresponding Bader charges. The Mulliken charges of Cu+ and O2− in Cu3AsO4 (Table 4) are consistent with those for Cu2O calculated via GGA (Cu: +0.33, O: −0.67).18) This outcome indicates that the ionicity of the bonding in Cu3AsO4 is common for Cu+-containing oxides. These results confirm that As–S bonding in Cu3AsS4 is covalent whereas As–O bonding in Cu3AsO4 is ionic. Therefore, Cu3AsO4 is assumed to be a complex oxide of Cu+ and As5+. Such difference in electronic structure between Cu3AsO4 and Cu3AsS4 is attributable to the fact that difference in the electronegativity of the As and O is larger than that between As and S (i.e., the Mulliken electronegativity values of O, S, and As are 2.75, 2.49, and 2.30, respectively).53) In metal nitrates, e.g., Zn(NO3)2 and Cd(NO3)2, the Zn 4s and Cd 5s states, respectively, do not contribute to the region just above the CBM; these regions are mostly composed of the antibonding states of N 2p and O 2p, exhibiting very small dispersion.54,55) This band structure is quite similar to that of Cu3AsS4. This coincidence also indicates that Cu3AsS4 is composed of Cu+ and AsS43−, similar to Zn(NO3)2 and Cd(NO3)2, which are composed of M2+ and NO3−.

Table 4 Mulliken charges of each atom of enargite-type Cu3AsO4 and Cu3AsS4 calculated via GGA+U with U = 4 eV.
The me* of Cu3AsS4 is quite large and cannot be determined by simple parabolic approximation (see Table 3). Therefore, high n-type conductivity is not expected for Cu3AsS4 even with appropriate doping, while this material experimentally exhibits p-type conduction without intentional doping.56) Indeed, n-type conduction has not been reported experimentally. Therefore, in research on Cu3AsS4 application in solar cells, devices consisting of a p-n heterojunction with n-type ZnS as the n-type layer have been considered.57)
Therefore, Cu3AsO4 has an advantage over Cu3AsS4 in terms of expected bipolar conduction by doping, which enables a p-n homojunction. A p-n homojunction is more desirable for photovoltaic conversion devices than a p-n heterojunction because it generally forms a high-quality interface with fewer defects, and as it exhibits ideal band alignment without band discontinuities such as spikes and cliffs.
3.4 Optical properties of enargite-type Cu3AsO4
Oxide semiconductors containing Cu+ are expected to have higher-energy VBMs yielding a narrower band gap than typical oxide semiconductors (band gap >3 eV). This is because the valence bands of Cu+-containing oxide semiconductors are mostly composed of the Cu 3d state, which has a higher energy than the O 2p state, as is the case for Cu2O (band gap: 2.1 eV)38) and β-CuGaO2 (1.5 eV).58) Table 5 summarizes the band gaps calculated for Cu3AsO4 and Cu3AsS4 together with the experimentally obtained value for Cu3AsS4. Underestimation of the band gap is a common feature of calculations using the GGA functional, even when U is introduced.59–61) Here, the calculated band gap of Cu3AsS4 was also underestimated. In the table, the calculated Cu3AsO4 band gap is slightly smaller than that of Cu3AsS4. As the experimentally determined Cu3AsS4 band gap is 1.28 eV,62) the Cu3AsO4 band gap is estimated to be 1–1.2 eV. This estimation is quite reasonable considering the Ag3AsS4 band gap of 1.9 eV,63) as Cu+-containing oxides have smaller band gaps than Ag+-containing oxides; for example, β-AgGaO2 (2.2 eV)64) and β-CuGaO2 (1.5 eV),58) and Ag3VO4 (2.2 eV)65) and Cu3VO4 (1.2 eV).66)

Table 5 Band gaps of enargite-type Cu3AsO4 and Cu3AsS4.
According to Shockley–Queisser theory, the 1–1.2-eV band gap is in the energy region corresponding to the highest theoretical maximum conversion efficiency of single-junction solar cells.16) Therefore, Cu3AsO4 is expected to be suitable for solar-cell applications in terms of its band gap, as is the case for Cu3AsS4.
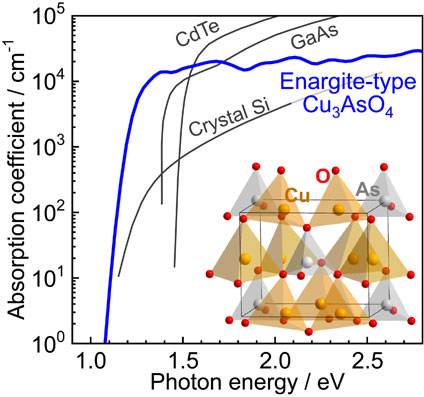
Figure 4 shows the calculated optical absorption spectra of enargite-type Cu3AsO4 and Cu3AsS4 with those of other absorber materials.67) The absorption coefficient of Cu3AsO4 reaches 104 cm−1 at photon energy of 1.3 eV, which corresponds to the value at which a thin film with 3-µm thickness absorbs >95% of incident light. This value is comparable to those of CdTe and GaAs above their direct band gaps, and these materials are already in practical use as thin-film solar cells. Although Cu3AsO4 is an indirect semiconductor, the absorption coefficient abruptly increases in the region immediately above the absorption edge. The energy difference between the band gap and photon energy where the absorption coefficient reaches 104 cm−1 is ∼0.3 eV for Cu3AsO4, which is only slightly larger than that for direct semiconductors such as CdTe, GaAs, and Cu3AsS4 (0.1–0.15 eV).68) This contrasts with the case of crystalline Si, for which the absorption coefficient reaches 104 cm−1 at 1.5 eV above its band gap. An abrupt absorption increase exhibited by Cu3AsO4 is because its smallest direct band gap is only 0.3 eV larger than its indirect band gap. Further, the region around the VBM is mostly composed of fully occupied Cu 3d states, yielding a large DOS in that region.
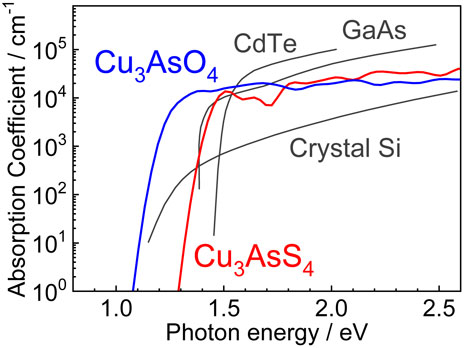
The above results indicate that Cu3AsO4 is a promising light-absorber material for thin-film solar cells, because it is expected to exhibit bipolar conduction by appropriate doping, a band gap of 1–1.2 eV suitable for solar cells, and a large optical absorption coefficient.
3.5 Possibility of enargite-type Cu3AsO4 formation
Figure 5 shows the results of a trial synthesis of Cu3AsO4 from Cu2O and As2O5 by a mechanical alloying technique. Note that this is one of the synthesis methods for obtaining a metastable phase. Following 30 min of mechanical alloying, metallic Cu and Cu3As2O8 formation was apparent in the sample, in addition to the unreacted raw materials (As2O5 and Cu2O) as shown in Fig. 5(a). An unidentified phase was also observed, corresponding to the diffractions marked by red arrows in the figure. These three unidentified diffractions can be attributed to those of stannite-type Cu3AsO4 (see Fig. 1(b)), which is one of the polymorphs of Cu3AsO4, as shown in Fig. 5(a).
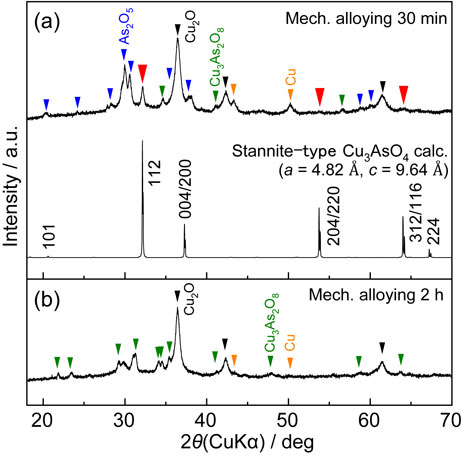
Although the formation enthalpy of stannite-type Cu3AsO4 is higher than that of enargite-type Cu3AsO4, the difference is very small (17 kJ mol−1). This outcome suggests that formation of the stannite-type rather than the enargite-type phase is possible by a mechanical alloying. Although the stannite-type Cu3AsO4 disappeared following 2 h of mechanical alloying (Fig. 5(b)), the formation of this material indicates the possibility of synthesis of enargite-type Cu3AsO4.