はじめに
歯周炎は歯周支持組織の慢性炎症であり,歯肉軟組織の歯への付着の喪失と歯槽骨吸収を特徴とし,プラーク細菌が歯周炎の病原性において不可欠な役割を演じていることが示されている。口腔内には500種類以上の細菌が生息し,これらの細菌は,共凝集や代謝物質の相互利用などの菌体間のさまざまな相互作用により互いの遺伝子発現や表現型に影響を及ぼしあい,口腔細菌叢を形成している。歯周組織が安定している状態では,口腔細菌叢と宿主の免疫防御機構との間で平衡状態(Controlled inflammation)が維持されているが,Red Complexとして知られるPorphyromonas gingivalis,Treponema denticolaやTannerella forsythiaなどのキーストーンとなる歯周病原細菌の存在や増加や1),免疫調節機構の変化,喫煙やストレス,加齢などの宿主体内の環境の変化はdysbiosisと呼ばれる細菌叢の乱れすなわち構成バランスの変化を誘導し,病原性の高い細菌叢へと変化させる。このような複数のリスク因子との複合的な作用による混合感染が最終的に歯周炎の発症・進行に関与すると考えられている2,3)。プラーク細菌による歯周組織破壊には宿主免疫応答が関係している。自然免疫による細菌に対する防御反応は,Pathogen-associated molecular patterns(PAMPs)と呼ばれる細菌に共通して存在する分子をToll-like receptors(TLRs)やNucleatide-binding and oligomerization domain(NOD)-like receptors(NLRs)などパターン分子認識受容体(PRRs)が細菌感染を検知することにより誘導される。TLRsやNLRsの刺激はnuclear factor-κB(NF-κB)とMitogen-activated protein kinases(MAPKs)を活性化し,インターロイキン(IL)-1βやIL-18などのさまざまなサイトカインやケモカインの遺伝子発現を誘導する。またマクロファージや樹状細胞のPRRsへの刺激による活性化は,T細胞への抗原提示を介した獲得免疫の活性化にも関与している。
IL-1βは単球/マクロファージから放出される炎症性サイトカインであり,その発現は歯周病患者の歯肉溝滲出液,唾液,歯肉において増加していることから歯周炎と強く関連していると推測されている4-6)。IL-1βは細菌感染に対する防御反応,炎症反応の調整に重要であり,破骨細胞の分化と活性化を促進することによって歯槽骨吸収を強く誘導する7)。翻訳されたIL-1βは生物学的活性を有せず,カスパーゼ1によりプロセッシングを受けることによりはじめて生物学的活性を発揮する(活性化型IL-1β)。カスパーゼ1による活性化の調節にはインフラマソームと呼ばれるタンパク複合体が関与しており,NLRファミリーに属するNOD-like receptor pyrin domain-containing protein 1(NLRP1),NLRP3,NLR family CARD domain-containing protein 4(NLRC4)に加えてAbsent in melanoma 2(AIM2)などのタンパクがインフラマソームを形成することが知られている8)。これらのインフラマソームの中でNLRP3インフラマソームは世界中で最もよく研究されており,その特性が明らかにされつつある。本稿ではNLRP3インフラマソームと歯周炎の関係について総説する。
NLRP3インフラマソーム
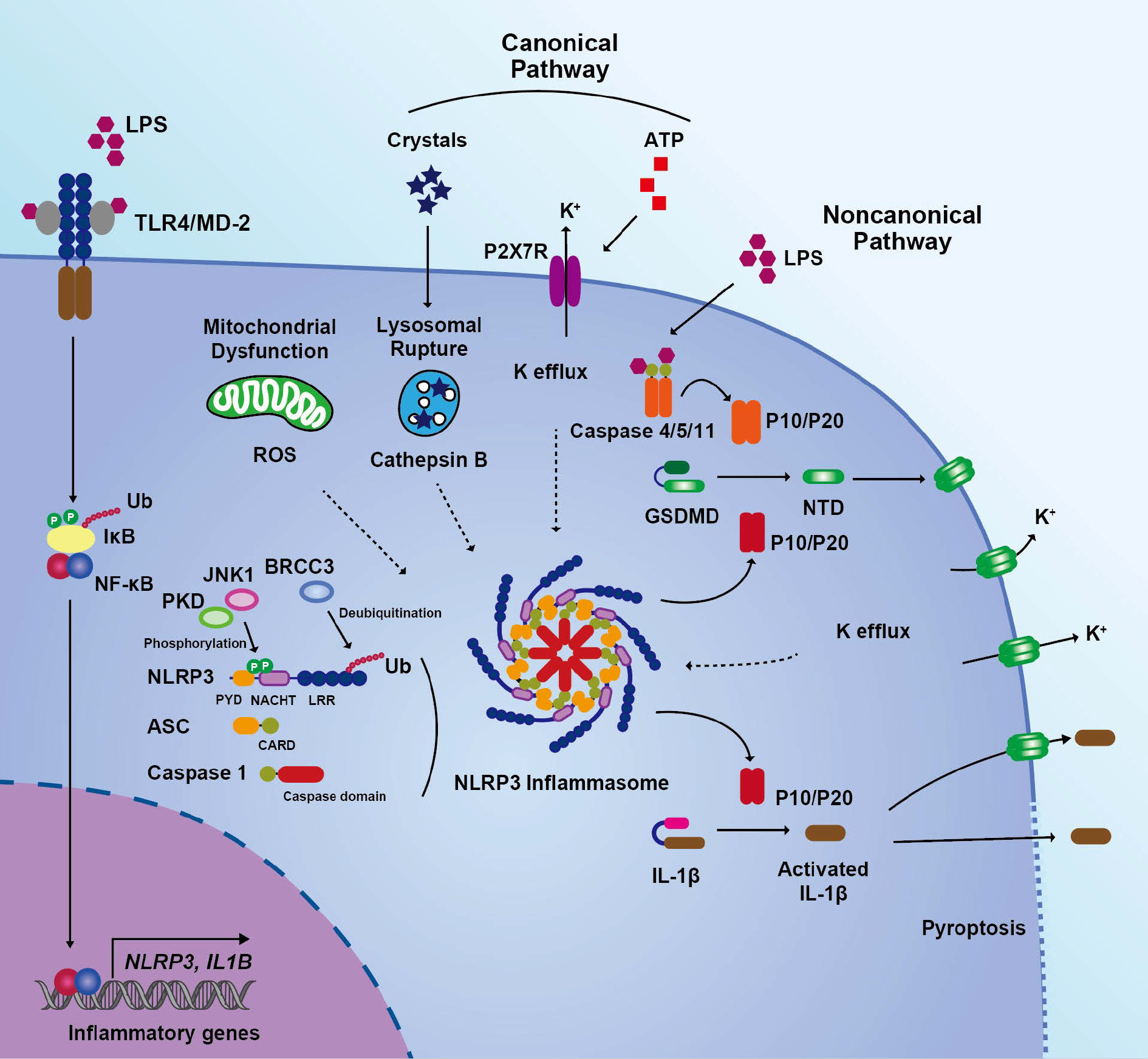
NLRP3はNLR familyに属する細胞質タンパクで,好中球,マクロファージなどの自然免疫細胞に発現し,2型糖尿病,痛風関節炎,アルツハイマー病などの炎症性疾患と関係しており,歯周病との関連性も推測されている。NLRP3はN末端からPyrinドメイン,中央にNACHTドメイン,そしてC末端のLeucin-rich repeat(LRR)より構成されている。NLRP3インフラマソーム形成は複雑に制御されており,プライミング過程(シグナル1)と活性化過程(シグナル2)からなる9,10)。シグナル1はNLRP3の遺伝子発現であり,TLRsやNLRsの刺激によるNF-κBの活性化はNLRP3のみならずカスパーゼ1やIL-1β遺伝子発現を刺激し,これらのタンパク発現を増加させる。シグナル2はNLRP3のリン酸化とユビキチン化による翻訳後調節そしてインフラマソーム形成の過程からなる。Jun N-terminal kinase(JNK1)やProtein kinase D(PKD)によるNLRP3のリン酸化はNLRP3活性化を促進し11,12),また脱ユビキチン化酵素のBRCC3によるLRRドメインの脱ユビキチン化はNLRP3のNACHTドメインを介したオリゴマー化と活性化を促進することが報告されている13)。オリゴマー化したNLRP3にNIMA-related kinase 7(NEK7)がスキャホールドタンパクとして作用することにより14),PYDドメインを介してアダプター分子のapoptosis-associated speck-like protein containing a CARD(ASC)と相互作用する。そしてASCのCARDドメインを介してエフェクター分子のカスパーゼ1と相互作用し,NLRP3インフラマソームと呼ばれる巨大分子を形成する(図1)。またカリウムイオンの流出,リソゾームの崩壊,ミトコンドリアの活性酸素種(ROS)を介した刺激がNLRP3インフラマソーム形成を誘導することが知られているがその機序はよくわかっていない。NLRP3インフラマソーム上で近接しあったカスパーゼ1は互いにプロセッシングすることにより強い活性を発揮する活性化型のP20/P10ヘテロテトラマーを形成し(活性化カスパーゼ1),活性化カスパーゼ1はIL-1βやIL-18のプロセッシングによる活性化に関与する一方,ガスダーミンファミリーのガスダーミンD(GSDMD)の切断にも関与している15)。切断されたGSDMDのN末端ドメインは細胞膜上で多量体化することにより細胞膜に小孔を形成し,このガスダーミン小孔は活性化IL-1βとIL-18のunconventionalな放出やイオン流出入を引き起こし,小孔数の増加により細胞膨隆,細胞死(ピロトーシス)の一連のイベントを引き起こす(図1)。この活性化経路はcanonical経路として知られている。放出された活性化IL-1β,活性化IL-18,DAMPsや酵素などの細胞内物質は周囲組織細胞の自然免疫応答や組織破壊を誘発し,炎症をさらに拡大させると考えられている。
またnoncanonical経路によるNLRP3インフラマソーム活性化が報告されている。カスパーゼ4,5(マウスカスパーゼ11)はグラム陰性細菌の細胞膜外膜に存在するリポ多糖(LPS)と直接的に結合する細胞内LPS受容体として機能することが報告された16,17)。これらのカスパーゼはLPSと結合すると高分子凝集物を形成し,カスパーゼ1と同様にセルフプロセッシングにより活性化される。活性化カスパーゼ4,5はその基質であるGSDMDを切断し,カリウムイオンの流出をおこすGSDMD小孔の形成を誘導し,この小孔を介したカリウムイオンの流出はNLRP3の活性化に導く(図1)。最近,LPSと結合するタンパクとしてNur77が同定され,Nur77はnoncanonical経路によるNLRP3インフラマソーム活性化に必須であること,さらにNLRP3と相互作用すること,そしてその相互作用にはLPSとミトコンドリアdsDNAの存在が必要であることが報告された。この論文において,LPS刺激時にGSDMDのN末端はミトコンドリア膜に結合し,ミトコンドリアDNAを細胞質に放出させることが示されている18)。
歯周炎とNLRP3インフラマソーム
1. 歯周組織でのNLRP3の発現
歯周炎歯肉におけるNLRP3インフラマソームの発現が組織学的に観察されている。NLRP3のタンパク発現は歯肉上皮細胞,固有層のリンパ球,線維芽細胞,血管内皮細胞に認められ,慢性歯周炎,侵襲性歯周炎で増加していること19),そして慢性歯周炎患者の歯肉上皮や固有層におけるNLRP3,ASC,カスパーゼ1の発現は健常者と比較して亢進していることが示されている6,20)。一方歯肉バイオプシー中のNLRP3インフラマソーム遺伝子転写物を解析した実験では,慢性歯周炎,侵襲性歯周炎歯肉でNLRP3とIL-1β mRNA発現が増加していることが示されているが6,19,21,22),ASC mRNA発現に関しては健常歯肉と比較して増加している報告6),差がない報告21),そして減少している報告22)と様々である。また歯周病患者血清と唾液中のNLRP3,ASC,IL-1βの濃度は健常者と比較して増加しており,NLRP3濃度は血清CRP濃度と相関すること,そしてNLRP3,ASCおよびIL-1β濃度は歯周炎の炎症パラメーター,歯周炎の広がり,重症度と相関することが報告されている23,24)。さらに慢性歯周炎歯肉中でGSDMDの切断がおきていることも示されている25)。これらの実験はNLRP3インフラマソームが歯周炎の発症や進行に関与していることを支持している。
2. 歯周炎モデルとNLRP3
実験動物を使用した実験的歯周炎においてNLRP3インフラマソームの役割が評価されている。NLRP3欠損マウスにおける結紮誘導歯周炎の歯槽骨吸収は,WTのものと比較して減少していること,そしてWTにおける歯槽骨吸収はNLRP3のインヒビターのMCC950の腹腔投与により抑制されることが報告されている26)。一方,熱殺菌したAggregatibacter actinomycetemcomitansの歯肉投与では,カスパーゼ1欠損マウスでは骨吸収が抑制されたが,NLRP3欠損マウスでは抑制されなかったことから,NLRP3インフラマソーム以外のインフラマソームの関与が示唆されている27)。我々はP. gingivalisとA. actinomycetemcomitansの菌体を加熱・超音波破砕処理し,それぞれラット歯肉に投与することにより実験的歯周炎モデルを作製し,菌体投与による炎症性細胞浸潤とTRAP陽性の破骨細胞形成を伴う歯槽骨吸収はNLRP3抑制活性を有するスルホニル尿素系の糖尿病治療薬のグリブリド(グリベンクラミドとしても知られる)の経口投与により抑制されることを報告した28)(図2)。P. gingivalis生菌を用いた感染による歯周炎モデルの実験では相反した報告がなされている。Yamaguchiらは,P. gingivalis生菌のマウス口腔内投与により誘導された歯槽骨吸収はNLRP3欠損マウスで減少していたことを示しているのに対して29),Cheatらは結紮線に加えてP. gingivalis生菌を感染させて歯周炎を誘発させたモデルにおいて,NLRP3欠損マウスはコントロールと比較して増加した歯槽骨吸収を示したことから,NLRP3の防御的な役割を報告している30)。NLRP3欠損マウスやASCの欠損マウスはLPS投与による致死には抵抗性を示すが,様々な細菌による感染においては菌の増殖を許容し,細菌数の増加の結果,臓器障害や個体死を引き起こすことが報告されていることから31-33),P. gingivalis生菌を用いた感染による歯周炎モデルの解釈には注意が必要となろう。
3. 歯周病原細菌とNLRP3(in vitro研究)
歯周病原細菌はNLRP3インフラマソームシグナリングに関与している。P. gingivalis,T. denticola,Prevotella nigrescent,A. actinomycetemcomitans,Fusobacterium nucleatumによるマクロファージや歯肉上皮細胞,歯肉線維芽細胞,歯根膜細胞のIL-1βの放出,カスパーゼ1の活性化や細胞死の誘導にはNLRP3が関与することが示されている。このことは上述したNLRP3インフラマソームと歯周病の病原性との関連性を支持している28,34-37)。これらの研究において,NLRP3の活性化には,細胞外ATP,カリウムイオンの流出,ROS,リソゾーム酵素のカテプシンBが関与することから,P2X7受容体,ミトコンドリアダメージ,リソゾーム崩壊が重要な役割を演じていることが示唆されている。複数の研究者はP. gingivalisやそのLPSはNLRP3に対する活性化能は低く,ATPで共刺激しないかぎりカスパーゼ1の活性化やIL-1βが放出されないことを報告している38-42)。P. gingivalisのタンパク分解酵素のジンジパインは,様々な細胞受容体や免疫グロブリン,補体などのタンパクにくわえ,活性化型のカスパーゼ1とIL-1βを分解することも知られている43)。我々は,さまざまの酵素などの影響を排除するため,熱処理後に超音波破砕したP. ginigivalis,A. actinomycetemcomitans,F. nucleatum,Escherichia coliでヒトTHP-1マクロファージを刺激した。どの菌体もカスパーゼ1そしてNLRP3依存性のIL-1βの放出を誘導したが,P. gingivalisは他の3菌種と比較して低いことを確認した28)。さらにin vitroにおける低濃度RANKLとM-CSFによりプライミングしたマウス骨髄マクロファージ(BMM)のE.coli LPS刺激による破骨細胞形成はNLRP3インヒビターのMCC950,カスパーゼ1インヒビターとIL-1 receptor antagonist(IL-1RA)により抑制されたことから,LPSによりNLRP3/カスパーゼ1を介した誘導された活性化L-1β産生がautocrineにBMMに作用して破骨細胞形成に関与していることが示された44)。
4. 咬合性外傷とNLRP3
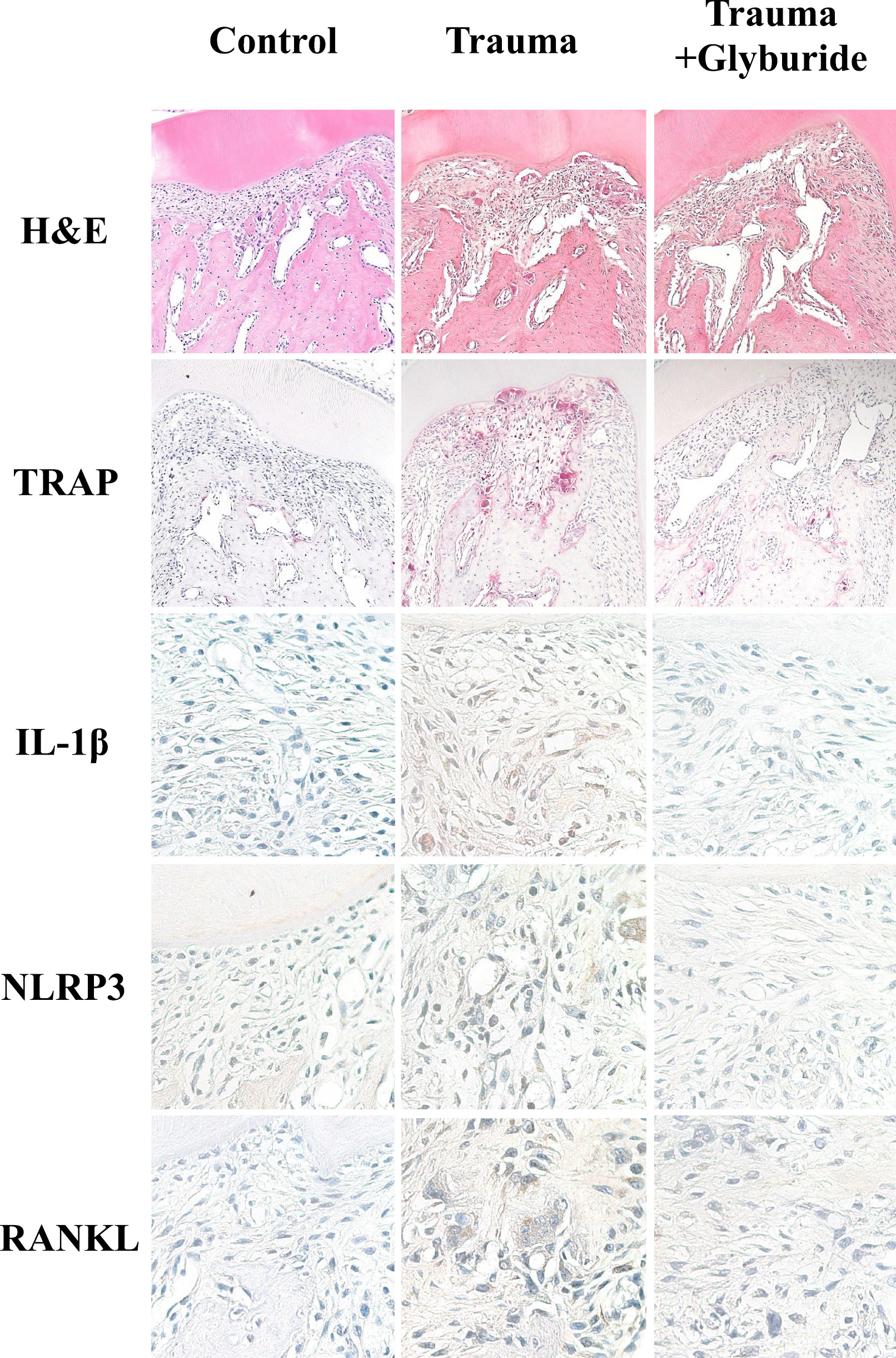
正常な咬合力は機械的刺激として,骨形成と骨吸収のバランスを調節することにより,歯根膜と歯槽骨のホメオスターシスに寄与している45,46)。しかし過剰な咬合力は咬合性外傷を引き起こし,歯根膜腔の拡大や歯槽骨の吸収を導くことが知られている47-49)。過剰な力による矯正治療では歯根膜細胞においてIL-1βの発現が増加すること,さらにIL-1βの刺激は歯根膜細胞のRANKLの発現や活性を増加させることから,我々は咬合性外傷による骨吸収におけるNLRP3インフラマソームの役割について検討した50)。ラット第一臼歯に外傷性咬合を与え,根分岐部における歯槽骨吸収を免疫組織学的に解析した。結果,咬合性外傷は分岐部の歯根膜腔の拡大と破骨細胞数を増加させ,歯根膜中のIL-1β,NLRP3,RANKL陽性細胞を増加させたが,NLRP3インヒビターのグリブリドを経口投与すると,これら全てが抑制された(図3)。このことから咬合性外傷における骨吸収にはNLRP3が関与していることが明らかになった。また同様にマウス矯正力によるTRAP陽性細胞の増加と歯の移動はNLRP3欠損マウスにおいて抑制されていることが報告されている51)。咬合性外傷においてどのような機序でNLRP3インフラマソームが活性化され,IL-1βが産生されるのかは明らかではないが,圧迫によって引き起こされる低酸素状態は歯根膜細胞にIL-1βのmRNAを増加させること52),また歯根膜細胞への機械的なストレスはATPの放出を刺激すること53),そして機械的なストレスによるIL-1βの発現はP2X7受容体(P2X7R)に依存していること54)が示されている。またHanら(2022)は圧迫力によるTHP-1マクロファージにおけるIL-1βの活性化にはcGAS-STING経路を介したNF-kBの活性化によるpro-IL-1βとNLRP3の発現増加とP2X7Rの活性化が重要であることを報告している51)。
最後に
前述したようにNLRP3は,IL-1βの活性化と放出とピロトーシスを介して炎症反応と骨吸収に関与し歯周病と密接に関連している。このことはNLRP3インフラマソームを標的とした歯周治療の可能性を示唆している。全身疾患のアルツハイマー病,多発性硬化症,脳卒中,2型糖尿病,非アルコール性肝炎や炎症性の腸疾患においても動物実験でNLRP3インヒビターの有効性が示され,また現在ヒトにおける多くの化合物による臨床治験が行われている55)。我々もグリブリドの経口投与がラット実験的歯周炎における歯肉の炎症性細胞浸潤と骨吸収そしてラット咬合性外傷による骨吸収を抑制することを報告したが,グリブリドは糖尿病治療薬であり,膵臓のβ細胞のATP感受性カリウムチャネル(Sur1-Kir6.2)を抑制し,インスリンの放出を促進し,血糖を低下させる作用も有する。将来,NLRP3に特異的に作用することにより副作用がなく,歯周炎に対し有効性を発揮する薬剤の開発が期待される。
今回の論文に関連して,開示すべき利益相反状態はありません。
References
- 1) Holt SC, Eversole JL: Treponema denticola, and Tannerella forsythia: the "red complex", a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000, 38: 72-122, 2005.
- 2) Darveau RP: Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol, 8: 481-490, 2010.
- 3) Hajishengallis G: Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol, 35: 3-11, 2014.
- 4) Tobon-Arroyave S, Jaramillo-Gonzalez P, Isaza-Guzman D: Correlation between salivary IL-1beta levels and periodontal clinical status. Arch Oral Biol, 53: 346-352, 2008.
- 5) Huang X, Yang X, Ni J, Xie B, Liu Y, Xuan D, Zhang J: Hyperglucose contributes to periodontitis: Involvement of the NLRP3 pathway by engaging the innate immunity of oral gingival epithelium. J Periodontol, 86: 327-335, 2015.
- 6) García-Hernández AL, Muñoz-Saavedra ÁE, González-Alva P, Moreno-Fierros L, Llamosas-Hernández FE, Cifuentes-Mendiola SE, Rubio-Infante N: Upregulation of proteins of the NLRP3 inflammasome in patients with periodontitis and uncontrolled type 2 diabetes. Oral Dis, 25: 596-608, 2019.
- 7) Schwartz Z, Goultschin J, Dean DD, Boyan BD: Mechanisms of alveolar bone destruction in periodontitis. Periodontol 2000, 14: 158-172, 1997.
- 8) Broz P, Dixit VM: Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol, 16: 407-420, 2016.
- 9) Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK: An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol, 18: 1141-1160, 2021.
- 10) Swanson KV, Deng M, Ting JPY: The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol, 19: 477-489, 2019.
- 11) Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, Liu X, Huang YJ, Cai H, Zhan XY, Han QY, Wang H, Chen Y, Li HY, Li AL, Zhang XM, Zhou T, Li T: NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell, 68: 185-197, 2017.
- 12) Zhang Z, Meszaros G, He WT, Xu Y, Magliarelli H de F, Mailly L, Mihlan M, Liu Y, Gámez MP, Goginashvili A, Pasquier A, Bielska O, Neven B, Quartier P, Aebersold R, Baumert TF, Georgel P, Han J, Ricci R: Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med, 214: 2671-2693, 2017.
- 13) Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J: Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell, 49: 331-338, 2013.
- 14) Xiao L, Magupalli VG, Wu H: Cryo-EM structures of the active NLRP3 inflammasome disc. Nature, 613: 595-600, 2023.
- 15) Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J: Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature, 535: 153-158, 2016.
- 16) Broz P, Monack DM: Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog, 9: e1003144, doi: 10.1371/journal.ppat.1003144, 2013.
- 17) Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynśki A, Forsberg LS, Carlson RW, Dixit VM: Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science, 341: 1246-1249, 2013.
- 18) Zhu F, Ma J, Li W, Liu Q, Qin X, Qian Y, Wang C, Zhang Y, Li Y, Jiang D, Wang S, Xia P: The orphan receptor Nur77 binds cytoplasmic LPS to activate the non-canonical NLRP3 inflammasome. Immunity, 56: 753-767, 2023.
- 19) Xue F, Shu R, Xie Y: The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT-PCR study and immunohistochemistry. Arch Oral Biol, 60: 948-958, 2015.
- 20) Ding PH, Yang MX, Wang NN, Jin LJ, Dong Y, Cai X, Chen LL: Porphyromonas gingivalis-induced NLRP3 inflammasome activation and its downstream interleukin-1β release depend on caspase-4. Front Microbiol, 11: 1881, doi: 10.3389/fmicb.2020.01881, 2020.
- 21) Bostanci N, Emingil G, Saygan B, Turkoglu O, Atilla G, Curtis MA, Belibasakis GN: Expression and regulation of the NALP 3 inflammasome complex in periodontal diseases. Clin Exp Immunol, 3: 415-422, 2009.
- 22) Aral K, Berdeli E, Cooper PR, Milward MR, Kapila Y, Karadede Ünal B, Aral CA, Berdeli A: Differential expression of inflammasome regulatory transcripts in periodontal disease. J Periodontol, 91: 606-616, 2020.
- 23) Isola G, Polizzi A, Santonocito S, Alibrandi A, Williams RC: Periodontitis activates the NLRP3 inflammasome in serum and saliva. J Periodontol, 93: 135-145, 2022.
- 24) Isaza-Guzmán DM, Medina-Piedrahíta VM, Gutiérrez-Henao C, Tobón-Arroyave SI: Salivary levels of NLRP3 inflammasome-related proteins as potential biomarkers of periodontal clinical status. J Periodontol, 88: 1329-1338, 2017.
- 25) Xia Y, Zhou K, Sun M, Shu R, Qian J, Xie Y: The miR-223-3p regulates pyroptosis through NLRP3-caspase 1-GSDMD signal axis in periodontitis. Inflammation, 44: 2531-2542, 2021.
- 26) Chen Y, Yang Q, Lv C, Chen Y, Zhao W, Li W, Chen H, Wang H, Sun W, Yuan H: NLRP3 regulates alveolar bone loss in ligature-induced periodontitis by promoting osteoclastic differentiation. Cell Prolif, 54: e12973, doi: 10.1111/cpr.12973, 2021.
- 27) Rocha FRG, Delitto AE, de Souza JAC, González-Maldonado LA, Wallet SM, Rossa Junior C: Relevance of caspase-1 and NLRP3 inflammasome on inflammatory bone resorption in a murine model of periodontitis. Sci Rep, 10: 7823, doi: 10.1038/s41598-020-64685-y, 2020.
- 28) Kawahara Y, Kaneko T, Yoshinaga Y, Arita Y, Nakamura K, Koga C, Yoshimura A, Sakagami R: Effects of Sulfonylureas on Periodontopathic Bacteria-Induced Inflammation. J Dent Res, 99: 830-838, 2020.
- 29) Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T: Regulation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated periodontal disease. Inflamm Res, 66: 59-65, 2017.
- 30) Cheat B, Torrens C, Foda A, Baroukh B, Sadoine J, Slimani L, Witko-Sarsat V, Huck O, Gosset M, Bouchet J: NLRP3 is involved in neutrophil mobilization in experimental periodontitis. Front Immunol, 13: 839929, doi: 103389/fimmu.2022.839929, 2022.
- 31) Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, Flavell RA: Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity, 24: 317-327, 2006.
- 32) Van Lieshout MH, Scicluna BP, Florquin S, Van Der Poll T: NLRP3 and ASC differentially affect the lung transcriptome during pneumococcal pneumonia. Am J Respir Cell Mol Biol, 50: 699-712, 2014.
- 33) Ceballos-Olvera I, Sahoo M, Miller MA, del Barrio L, Re F: Inflammasome-dependent pyroptosis and IL-18 protect against burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog, 7: e1002452, doi: 10.1371/journal.ppat.1002452, 2011.
- 34) Huang MT, Taxman DJ, Holley-Guthrie EA, Moore CB, Willingham SB, Madden V, Parsons RK, Featherstone GL, Arnold RR, O'Connor BP, Ting JP: Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol, 182: 2395-2404, 2009.
- 35) Jun HK, Lee SH, Lee HR, Choi BK: Integrin α5β1 activates the NLRP3 inflammasome by direct interaction with a bacterial surface protein. Immunity, 36: 755-768, 2012.
- 36) Shenker BJ, Ojcius DM, Walker LP, Zekavat A, Scuron MD, Boesze-Battaglia K: Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect Immun, 83: 1487-1496, 2015.
- 37) Jang HM, Park JY, Lee YJ, Kang MJ, Jo SG, Jeong YJ, Cho NP, Cho SD, Kim DJ, Park JH: TLR2 and the NLRP3 inflammasome mediate Il-1β production in prevotella nigrescens-infected dendritic cells. Int J Med Sci, 18: 432-440, 2021.
- 38) Ramos-Junior ES, Morandini AC, Almeida-Da-Silva CLC, Franco EJ, Potempa J, Nguyen KA, Oliveira AC, Zamboni DS, Ojcius DM, Scharfstein J, Coutinho-Silva R: A Dual Role for P2X7 Receptor during Porphyromonas gingivalis Infection. J Dent Res, 94: 1233-1242, 2015.
- 39) De Andrade KQ, Almeida-da-Silva CLC, Ojcius DM, Coutinho-Silva R: Differential involvement of the canonical and noncanonical inflammasomes in the immune response against infection by the periodontal bacteria Porphyromonas gingivalis and Fusobacterium nucleatum. Curr Res Microb Sci, 2: 100023, doi: 10.1016/j.crmicr.2021.100023, 2021.
- 40) Xu S, Zhou Q, Fan C, Zhao H, Wang Y, Qiu X, Yang K, Ji Q: Doxycycline inhibits Nacht Leucine-rich repeat protein 3 inflammasome activation and interleukin-1β production induced by Porphyromonas gingivalis-lipopolysaccharide and adenosine triphosphate in human gingival fibroblasts. Arch Oral Biol, 107: 104514, doi: 10.1016/j.archoralbio.2019.104514, 2019.
- 41) Guo W, Wang P, Liu Z, Yang P, Ye P: The activation of pyrin domain-containing-3 inflammasome depends on lipopolysaccharide from Porphyromonas gingivalis and extracellular adenosine triphosphate in cultured oral epithelial cells. BMC Oral Health, 15: 133, doi: 10.1186/s12903-015-6, 2015.
- 42) Yilmaz Ö, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM: ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol, 12: 188-198, 2010.
- 43) Okano T, Ashida H, Suzuki S, Shoji M, Nakayama K, Suzuki T: Porphyromonas gingivalis triggers NLRP3-mediated inflammasome activation in macrophages in a bacterial gingipains-independent manner. Eur J Immunol, 48: 1965-1974, 2018.
- 44) Alam MI, Mae M, Farhana F, Oohira M, Yamashita Y, Ozaki Y, Sakai E, Yoshimura A: NLRP3 inflammasome negatively regulates RANKL-induced osteoclastogenesis of mouse bone marrow macrophages but positively regulates it in the presence of lipopolysaccharides. Int J Mol Sci, 23: 6096, doi: 10.3390/ijms23116096, 2022.
- 45) Tsuji K, Uno K, Zhang GX, Tamura M: Periodontal ligament cells under intermittent tensile stress regulate mRNA expression of osteoprotegerin and tissue inhibitor of matrix metalloprotease-1 and -2. J Bone Miner Metab, 22: 94-103, 2004.
- 46) Wang JHC, Thampatty BP, Lin JS, Im HJ: Mechanoregulation of gene expression in fibroblasts. Gene, 391: 1-15, 2007.
- 47) Yoshinaga Y, Ukai T, Abe Y, Hara Y: Expression of receptor activator of nuclear factor kappa B ligand relates to inflammatory bone resorption, with or without occlusal trauma, in rats. J Periodontal Res, 42: 402-409, 2007.
- 48) Walker CG, Ito Y, Dangaria S, Luan X, Diekwisch TGH: RANKL, osteopontin, and osteoclast homeostasis in a hyperocclusion mouse model. Eur J Oral Sci, 116: 312-318, 2008.
- 49) Tsuzuki T, Kajiya H, T-Goto K, Tsutsumi T, Nemoto T, Okabe K, Takahashi Y: Hyperocclusion stimulates the expression of collagen type XII in periodontal ligament. Arch Oral Biol, 66: 86-91, 2016.
- 50) Arita Y, Yoshinaga Y, Kaneko T, Kawahara Y, Nakamura K, Ohgi K, Arita S, Ryu T, Takase M, Sakagami R: Glyburide inhibits the bone resorption induced by traumatic occlusion in rats. J Periodontal Res, 55: 464-471, 2020.
- 51) Han Y, Yang Q, Huang Y, Gao P, Jia L, Zheng Y, Li W: Compressive force regulates orthodontic tooth movement via activating the NLRP3 inflammasome. FASEB J, 36: e22627, doi: 10.1096/fj.202200447RR, 2022.
- 52) Motohira H, Hayashi J, Tatsumi J, Tajima M, Sakagami H, Shin K: Hypoxia and Reoxygenation Augment Bone-Resorbing Factor Production From Human Periodontal Ligament Cells. J Periodontol, 78: 1803-1809, 2007.
- 53) Ito M, Arakawa T, Okayama M, Shitara A, Mizoguchi I, Takuma T: Gravity loading induces adenosine triphosphate release and phosphorylation of extracellular signal-regulated kinases in human periodontal ligament cells. J Investig Clin Dent, 5: 266-274, 2014.
- 54) Kanjanamekanant K, Luckprom P, Pavasant P: Mechanical stress-induced interleukin-1beta expression through adenosine triphosphate/P2X7 receptor activation in human periodontal ligament cells. J Periodontal Res, 48: 169-176, 2013.
- 55) Coll RC, Schroder K, Pelegrín P: NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci, 43: 653-668, 2022.