受賞総説
パイロトーシスを標的とする新たな機序で働く抗炎症薬の開発
2023 年 143 巻 12 号 p. 997-1003
詳細
2023 年 143 巻 12 号 p. 997-1003
Programmed cell death plays various physiological roles, one of which is an immune response that protects the body from infectious pathogens such as bacteria and viruses. Pathogen infection causes dysfunction of cellular organelles, such as mitochondria and lysosomes, triggering stress signals that induce programmed cell death. In some cases, cell death coincides with intracellular inflammatory cytokine release. Such programmed cell death, accompanied by the induction of inflammatory responses, is called pyroptosis, which inhibits pathogen proliferation within cells and attracts leukocytes that eliminate the pathogens, thereby preventing infection spread. Additionally, pyroptosis can be induced by noninfectious stimuli such as drugs, pollutants, and nutrients, resulting in severe inflammatory disease. Therefore, the development of effective anti-inflammatory drugs that prevent pyroptosis based on the understanding of the mechanisms responsible for its induction is an urgent requirement. This review provides an overview of the non-infectious inflammatory response caused by pyroptosis and the development of new anti-inflammatory drugs that target organelles to prevent pyroptosis to treat relevant inflammatory diseases.
私たちの身体を構成する細胞は,その寿命とは無関係に細胞死する場合がある.細胞死はプログラム細胞死と非プログラム細胞死に分けられ,前者はアポトーシス,ネクロプトーシス,パイロトーシスなどが,後者ではネクローシスが挙げられる.発生の過程で不要になった細胞や,ストレスによる異常を抱えた細胞などは,身体に残ると望ましくないため,それらを能動的に除去するためにプログラム細胞死が誘導される.一方で,ネクローシスは,ストレスにさらされた細胞が陥る分子機序のない受動的な細胞死であるが,細胞が死滅する際に細胞内成分が漏出して免疫を活性化し,炎症応答を誘発してしまうことがよく知られている.重要なことに,プログラム細胞死についても,不適切な状況,あるいは過剰な頻度で誘導されると,炎症応答の引き金になる場合がある.筆者はこれまでに細胞死–炎症応答–疾患形成のつながりを免疫学の観点から研究し,治療戦略が十分に確立されていなかった炎症性疾患に対して新たな薬物療法を提案してきた.1,2)
私たちの身体に備わっている免疫は,異物,とりわけ病原体を排除する重要な防御機能であり,自然免疫と獲得免疫に大別される.そのうち,自然免疫は感染初期の防御応答において重要な役割を果たしている.自然免疫は,病原体を構成する特有の成分や,病原体感染を起因とするオルガネラの機能不全によって生じるストレスシグナルを,パターン認識受容体(pattern recognition receptors: PRRs)が認識することで活性化する.感染を感知したPRRsは,炎症性サイトカイン,I型インターフェロン,ケモカインなどの発現を誘導し,それぞれが白血球を活性化又は誘致することで,病原体排除のための炎症応答を速やかに惹起する.3,4)また,PRRsによっては細胞死を誘導する場合もある.細胞内感染する細菌やウイルスは細胞の栄養や機能を利用して増殖する.したがって,病原体感染に対して細胞死を誘導することは,病原体の増殖の場を奪うことにつながるため,結果として感染防御としての役割を果たす.前述のように細胞死には様々な様式があるが,PRRsが誘導する細胞死のうち,近年ではパイロトーシスが注目されており,関連する分子機序や,感染等に伴う病態形成への係わりについての研究が今も盛んに報告されている.パイロトーシス(pyroptosis)は,ギリシャ語で炎や熱を意味する「pyro」と,落ちるの意味で合目的な死を表す「ptosis」という2つの語をつなげて命名されたもので,5)細胞死する際に細胞が膨らんで破裂するとともに,細胞内から炎症性サイトカインであるInterleukin(IL)-1βやIL-18などの放出が起き,能動的に炎症応答を起こすことが特徴として知られている.
2000年代からパイロトーシスの解析が加速度的に進んだことと相まって,パイロトーシスは感染とは無関係の疾患の病態形成にも深く係わるという報告が相次いだ.PRRsはその名前の通り,類似性が高い一連の分子群をパターン認識するため,獲得免疫系を担うT細胞やB細胞が持つ異物認識機構(抗原受容体)と比べると特異性が低く,病原体以外の外的要因によっても活性化してしまう場合がある.そのようにして誘導される非感染性の炎症応答は「自然炎症」と呼ばれる.6)このように自然免疫は正負の両面性を持ち,様々な疾患の発症に深く係わっているため,その制御機構の解明や,その理解に基づく予防・治療法の開発は,重要な研究課題だと考えられている.とくにパイロトーシスが引き起こす自然炎症は,わが国を始めとする先進国において大きな問題となっている生活習慣病,薬剤副作用,環境性肺疾患などの様々な疾患の要因となっており,重要な治療標的として注目されている.
パイロトーシスがプログラム細胞死の様式の1つとして認められるより以前,細胞死の様式は主にアポトーシスとネクローシスしか具体的に定義されていなかった.パイロトーシスが公的に初めて認識されたのは1990年代後半のArturo Zychlinskyらの研究論文であり,赤痢菌やサルモネラ菌に感染したマクロファージがCaspase-1に依存して細胞死を起こすことが報告された.7,8)後に解析が進み,Caspase-1依存的かつCaspase-3非依存的に細胞膜の破裂が起こるとともに,発熱性の炎症性サイトカインであるIL-1β(この働きにちなんで,当該分子はpyrogenと言われる)の放出を起こすことを特徴とする新たな細胞死として定義された.
病原体感染を感知し,パイロトーシスを誘導するPRRsとしてNOD様受容体(nucleotide-binding oligomerization domain-like receptor: NLR)ファミリーに属する分子群が知られており,Caspase-1とともにインフラマソームと呼ばれる複合体を形成して,下流の分子群を活性化させる.NLRファミリーは,NOD1に存在するNACHTドメインと相同性の高いアミノ酸領域を有する.3) NLRファミリーは細胞内に侵入した病原体を感知する役割を果たしており,マクロファージ,単球,樹状細胞などのミエロイド系細胞において主に機能する.NLRファミリーの中でも,感染防御と自然炎症の両方に深い係わりを持つ分子として,nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3(NLRP3,別名としてNALP3, CIAS1, Cryopyrinなど)が知られている.NLRP3は,アダプター分子であるapoptosis-associated speck-like protein containing a caspase recruitment domain(ASC),Caspase-1とともにインフラマソームを形成する(Fig. 1).3,4)インフラマソームが活性化すると,Caspase-1が細胞内に存在するIL-1βやIL-18の前駆体を切断して活性化型への成熟化を促す.さらに,Caspase-1はGasdermin Dを切断し,そのN末端断片が多量体化することで細胞膜に小孔が形成される.これによりIL-1βやIL-18が小孔から細胞外へと放出されるとともに,小孔から細胞外液が流入して細胞が膨潤し,ひいては細胞が破裂して死に至る.
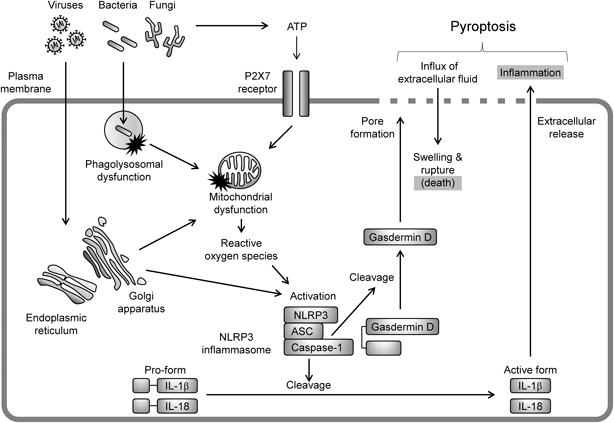
Pathogens damage organelles, resulting in their dysfunction. Organelle dysfunction induces NLRP3 inflammasome activation. Upon inflammasome activation, caspase-1 cleaves the precursors of IL-1β and IL-18 into their active forms. Furthermore, caspase-1 cleaves gasdermin D to convert it to its active form, which in turn forms pores in the plasma membranes. An increase in plasma membrane permeability promotes not only the extracellular release of IL-1β and IL-18 but also the influx of extracellular fluid, leading to cell swelling, rupture, and death (pyroptosis).
NLRP3インフラマソームは,黄色ブドウ球菌,A群連鎖球菌,B群連鎖球菌などの細菌,カンジダなどの真菌,インフルエンザウイルスなどのウイルスといった,様々な病原体を認識して活性化する.3,4) NLRP3インフラマソームが細菌,真菌,ウイルスという広範囲の病原体の感染を感知できるのは,これらが感染した際に誘導される種々の細胞小器官(オルガネラ)の機能不全をNLRP3が感知するためである.例として,細菌や真菌を免疫細胞が貪食した際に,それらを取り込んだファゴソームがCathepsinなどの酵素を内包するリソソームと融合(ファゴリソソームになる)して消化しようとするが,菌はそこから逃れるために膜溶解性毒素を産生してファゴリソソーム膜を損傷させる.損傷したファゴリソソームから内容物が漏れ出すと,ミトコンドリアなど他のオルガネラの機能不全を招く.また,ウイルス感染の際には,小胞体やゴルジ体からカルシウムイオンや水素イオンが流出する,あるいはウイルスタンパク質が発現するなどして,ミトコンドリアの機能不全や直接的なNLRP3インフラマソームの活性化が起きる.また,細菌の細胞外アデノシン三リン酸(adenosine triphosphate: ATP)によるP2X7受容体の活性化,膜溶解性毒素による細胞膜損傷,ファゴリソソームの損傷によって細胞質に漏出するCathepsinによる細胞膜刺激などを原因として,細胞膜の透過性が高まり,カリウムイオンの細胞外流出が起きた場合でもミトコンドリアの機能不全が起きる.機能不全を起こしたミトコンドリアからは活性酸素種が産生され,NLRP3インフラマソームを活性化させる.近年では,グラム陰性菌の感染時に,その膜成分であるリポ多糖が細胞質のCaspase-4/5(マウスではCaspase-11)に結合し,上記とは全く別の経路でNLRP3インフラマソームを活性化することも知られている.9)
前述のように,NLRP3は病原体感染に伴い機能不全を起こしたオルガネラから生じるストレスシグナルを感知するものであり,病原体の成分を認識する訳ではない.そのため,非感染性の外的刺激であっても,病原体感染と同様のストレスシグナルを生じさせる場合,NLRP3インフラマソームが活性化してしまう.3)例えば,過栄養により体内に蓄積する尿酸塩やコレステロールの結晶,環境汚染物質である黄砂やPM2.5,工業用マテリアルであるアスベストやカーボンナノチューブ,加齢に伴い蓄積するアミロイド-βなどは,免疫刺激性の微粒子として,それぞれ痛風,動脈硬化症,塵肺,慢性閉塞性肺疾患,中皮腫,アルツハイマー病の発症要因となる.これらの微粒子をマクロファージなどの免疫細胞が貧食すると,微粒子が持つ鋭利な構造や特殊な表面性状によってファゴリソソームの膜が損傷するが,病原体感染による損傷と区別がつかないため,NLRP3インフラマソームが誤って活性化する.これによって起きるパイロトーシスを介して炎症応答が誘導されても,原因となった微粒子は病原体のように除去されず,微粒子の残存あるいは度重なる暴露によって炎症応答が慢性化すると,組織障害を引き起こしてしまう.また,物理的あるいは化学的なストレスにより細胞が損傷することで細胞内から漏出したATPがNLRP3インフラマソームを活性化させ,炎症応答を介して組織障害を招く場合もある.このような現象はアセトアミノフェンやスタチンなどの薬剤副作用に深く係わると考えられている.このように,NLRP3インフラマソーム依存的なパイロトーシスは様々な非感染性の疾患の発症に係わっており,有力な治療標的と考えられている.
筆者が属する研究グループは,NLRP3インフラマソームの制御機構に働きかける化合物を探索する中で,痛風発作治療薬の一つとして頻用されるコルヒチンが,尿酸塩結晶によるNLRP3インフラマソームの活性化を抑えることを観察した.10)コルヒチンの公知の作用として,微小管の主要タンパク質であるチューブリンに結合して重合を阻害し,微小管の形成を妨げることがわかっている.オルガネラの1つである微小管は,α及びβチューブリンにより構成される管状の構造体であり,細胞骨格の維持や,ミトコンドリアや小胞体などの他のオルガネラの局在を規定する働きを持っている.尿酸塩結晶を貧食したマクロファージでは,ファゴリソソーム膜の損傷に応じて,ミトコンドリアが微小管上をダイニン依存的に移動し,小胞体へと近接する.NLRP3は小胞体上に,アダプター分子であるASCはミトコンドリア上に主に局在しており,両オルガネラが近接することにより,NLRP3インフラマソームの形成が促される.微小管を介した小胞体とミトコンドリアの近接は,αチューブリンのアセチル化修飾に依存する.11)コルヒチンは微小管重合を阻害するため,結果として上記の機構を介したNLRP3インフラマソームの形成が妨げられ,尿酸塩結晶による炎症が緩和される.一方で,コルヒチンは微小管重合を阻害するがゆえに細胞毒性が強いという注意点がある.そこで細胞毒性が低い化合物を探索したところ,食物に含まれるポリフェノールとして著名なレスベラトロールがαチューブリンのアセチル化を阻害し,NLRP3インフラマソームの活性化を抑制することを見い出した.12)実のところ,NLRP3インフラマソームや,その制御を受ける分子を直接の標的とする薬剤について,精力的に開発・試験が進められている.例えば,IL-1βの働きを抑制する目的で,抗ヒトIL-1βモノクローナル抗体であるCanakinumabやGevokizumab,ヒトIL-1受容体拮抗薬であるAnakinra, IL-1をトラップするRilonaceptなどのタンパク質製剤が,動物試験や臨床試験において,痛風関節炎に対して一定の抑制効果を上げている.13,14)また,NLRP3を直接阻害する薬剤としてMCC950を筆頭にいくつかの低分子化合物の動物試験や臨床試験も進んでいる.15)このように,オルガネラを標的とする抗炎症薬はこれまであまり注目されていなかったものの,NLRP3インフラマソームの活性化を抑えるという目的において合理的であり,パイロトーシスを起因とする炎症性疾患に対する新たな予防・治療薬になることが期待される.
最近の筆者の研究として,外的刺激によるオルガネラの機能不全を抑える抗炎症薬の開発を進めている.オルガネラの機能不全は,NLRP3インフラマソームの活性化以外にも,下流で炎症応答を誘導することが示唆されている.例えば,ミトコンドリアの機能不全は活性酸素種やミトコンドリアDNAの産生を誘発し,Toll様受容体やcyclic GMP-AMP synthaseといった他のPRRsの活性化を介して炎症応答を増幅する.16,17)すなわち,NLRP3インフラマソームやその下流で働く炎症性サイトカインなどを標的とするよりも,その上流においてミトコンドリアなどのオルガネラの機能不全を抑制する方が,炎症応答の誘導を広範囲に抑えられる可能性がある.筆者らは,放線菌由来の抗生物質nanaomycin(NNM)-Eが,外的刺激によって誘導されるミトコンドリアの機能不全を抑制し,NLRP3インフラマソームの活性化とパイロトーシスを防ぐことを見い出した(Fig. 2).18) NNM-Eと同じファミリーに属するNNM-Aは,牛の皮膚真菌病に対する外用薬として使用されており,NNM-Eと同等にNLRP3インフラマソームの活性化を抑制することを見い出したが,強い細胞毒性があった.その点で,NNM-Eは細胞毒性が非常に低く,望ましく思われた.NNM-Aが外用薬として利用されていることも踏まえ,生体におけるNNM-Eの抗炎症効果は乾癬様皮膚炎モデルを用いて評価した.抗ウイルス薬であるイミキモド(imiquimod: IMQ)を実験動物の皮膚に反復塗布すると,乾癬様の皮膚炎が起きることが古くから知られている.IMQはヒトでも外用時に皮膚炎症状を引き起こす場合がある.重要なことに,IMQはミトコンドリアの機能不全を誘導してNLRP3インフラマソームを活性化する.19)また,NLRP3はヒトの乾癬の感受性遺伝子であること,20)またIMQ誘導性乾癬様皮膚炎モデルもNLRP3依存的であることが支持されている.21)マウスにNNM-Eを塗布したところ,IMQ誘導性の乾癬様皮膚炎が顕著に抑えられた.乾癬をはじめ,NLRP3インフラマソーム依存的なパイロトーシスを起因とする炎症性疾患に対する薬剤療法の開発に,NNM-Eが役立てられることが望まれる.
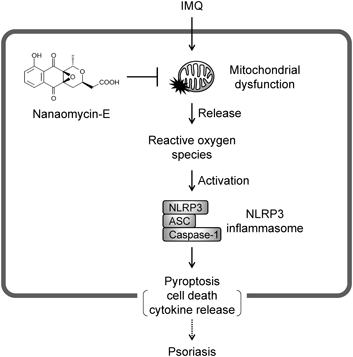
Nanaomycin E inhibits imiquimod (IMQ)-induced activation of the NLRP3 inflammasome and pyroptosis by preventing mitochondrial dysfunction.
近年の研究により,NLRP3インフラマソームを活性化する外的刺激のうち,微粒子が誘導する自然炎症は,NLRP3インフラマソームやその下流で働く炎症性サイトカインを阻害するだけでは,十分に抑えられないことがわかってきた.その理由として,微粒子により誘導される細胞死の実行シグナルはNLRP3インフラマソームのみに依存していないこと,更にNLRP3インフラマソームを介さずとも活性を示す炎症誘発分子がそのような細胞死に伴って放出されることがわかっている(Fig. 3A).よく研究されている例として,IL-1βと同じ受容体に作用するIL-1αが微粒子による刺激でNLRP3非依存的に放出され,痛風,動脈硬化症,肺の急性障害やアレルギー性炎症に深く係わることが示唆されている.22–25)実際に,マウスのマクロファージや樹状細胞を微粒子で刺激すると,NLRP3非依存的にIL-1αが放出される.22,24,26) IL-1αは多くの細胞の核内に常時発現しており,細胞がストレスを受けて死滅し,生体膜が崩壊すると,細胞外に漏出して炎症応答を誘発する.また,IL-1αはIL-1βとは異なり,Caspsase-1による切断を受けずとも活性を示す.一方で,NLRP3インフラマソームの活性化に貪食作用を必要としないATPやIMQのような刺激の場合は,IL-1βとIL-1αの放出はいずれもNLRP3に依存する(Fig. 3B).19,24,27)この理由として,微粒子によるファゴリソソームの損傷が起点となり,NLRP3インフラマソーム以外にも細胞死の実行シグナルが送られていることがわかってきている.28)
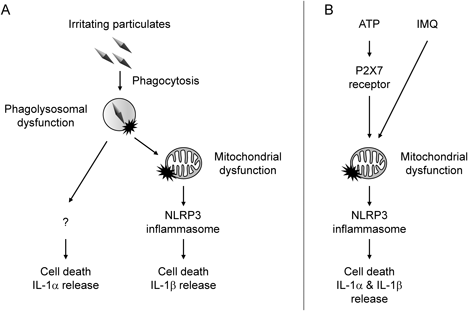
A: Particulate-induced phagolysosomal dysfunction induces NLRP3 inflammasome activation through mitochondrial dysfunction, inducing IL-1β release and cell death. Phagolysosomal dysfunction also induces IL-1α release and cell death via NLRP3-independent unidentified pathways. B: ATP or IMQ cause mitochondrial dysfunction and induce the release of IL-1α and IL-1β and cell death by activating the NLRP3 inflammasome.
微粒子によるパイロトーシスを防ぐ手段の1つとして,免疫細胞による微粒子の貪食を阻害し,ファゴリソソームの損傷を未然に防ぐことが考えられる.これまでに微粒子の認識に働く受容体としてSR-A1, SR-B1, Tim4, Siglec-14などが報告されており,それぞれの働きを阻害する抗体や薬剤によって微粒子の貪食が抑えられることが示されている.29–32)
一方で筆者らは,貪食作用には影響せず,微粒子によるファゴリソソームの損傷を抑える化合物として,延命草という生薬の成分であるOridoninを同定した.26) Oridoninで処理したマウスマクロファージでは,微粒子によるファゴリソソームの損傷が抑えられる結果として,NLRP3インフラマソーム依存的なIL-1βの放出だけでなく,NLRP3インフラマソーム非依存的な細胞死に伴うIL-1αの放出が顕著に抑えられた(Fig. 4).さらに,マウスに微粒子を吸引させて急性肺傷害を誘導した際に,Oridoninを投与しておくと,肺組織の炎症が顕著に抑えられることを示した.このように,オルガネラの機能不全を抑える抗炎症薬というコンセプトは,微粒子により誘導されるパイロトーシスに起因する自然炎症に対する治療薬の開発においても有効であろうと期待される.
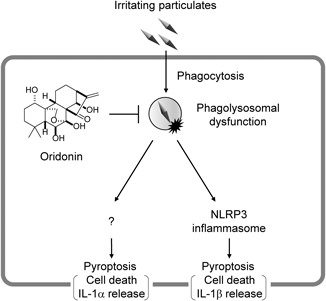
Oridonin inhibits particulate-induced pyroptosis by preventing phagolysosomal dysfunction, resulting in the inhibition of the release of both IL-1α and IL-1β.
自然炎症を誘発するパイロトーシスについて,NLRP3インフラマソームに依存する経路に関しては一定の理解が得られており,それに基づき開発された薬剤の一部は既に臨床に応用されるまでに至っている.一方で,微粒子により誘導されるパイロトーシスについては,誘導に係わる分子機序や,病態形成との係わりについて,解明すべき点が多く残されている.オルガネラは古典的に知られているような細胞の生命活動を維持する役割に加え,炎症シグナルを媒介することで細胞の外にも働きかけることが新たにわかってきており,非常に興味深い.今後,オルガネラを解析対象として,細胞死や免疫に関する基礎研究を続けることで,効果的な疾患治療薬の開発,ひいては健康社会の発展に貢献していきたい.
本稿に係わる主な研究は,大阪大学 大学院薬学研究科 生体応答制御学分野において実施されたものであり,齊藤達哉教授,髙濵充寛特任助教をはじめとする研究室のすべての方々に厚く御礼を申し上げます.
開示すべき利益相反はない.
本総説は,2022年度日本薬学会関西支部奨励賞(生物系薬学)の受賞を記念して記述したものである.