
Volume 28, Issue 4
Displaying 1-8 of 8 articles from this issue
- |<
- <
- 1
- >
- >|
Review (KSPE-JSPE Plenary Lecture)
-
2019 Volume 28 Issue 4 Pages 97-103
Published: 2019
Released on J-STAGE: October 19, 2019
Download PDF (470K)
Original Article
-
2019 Volume 28 Issue 4 Pages 105-112
Published: 2019
Released on J-STAGE: October 19, 2019
 Download PDF (537K)
Download PDF (537K) -
2019 Volume 28 Issue 4 Pages 113-125
Published: 2019
Released on J-STAGE: October 19, 2019
 Download PDF (753K)
Download PDF (753K) -
2019 Volume 28 Issue 4 Pages 127-133
Published: 2019
Released on J-STAGE: October 19, 2019
 Download PDF (586K)
Download PDF (586K)
Case Report
-
2019 Volume 28 Issue 4 Pages 135-138
Published: 2019
Released on J-STAGE: October 19, 2019
Download PDF (945K) -
2019 Volume 28 Issue 4 Pages 139-145
Published: 2019
Released on J-STAGE: October 19, 2019
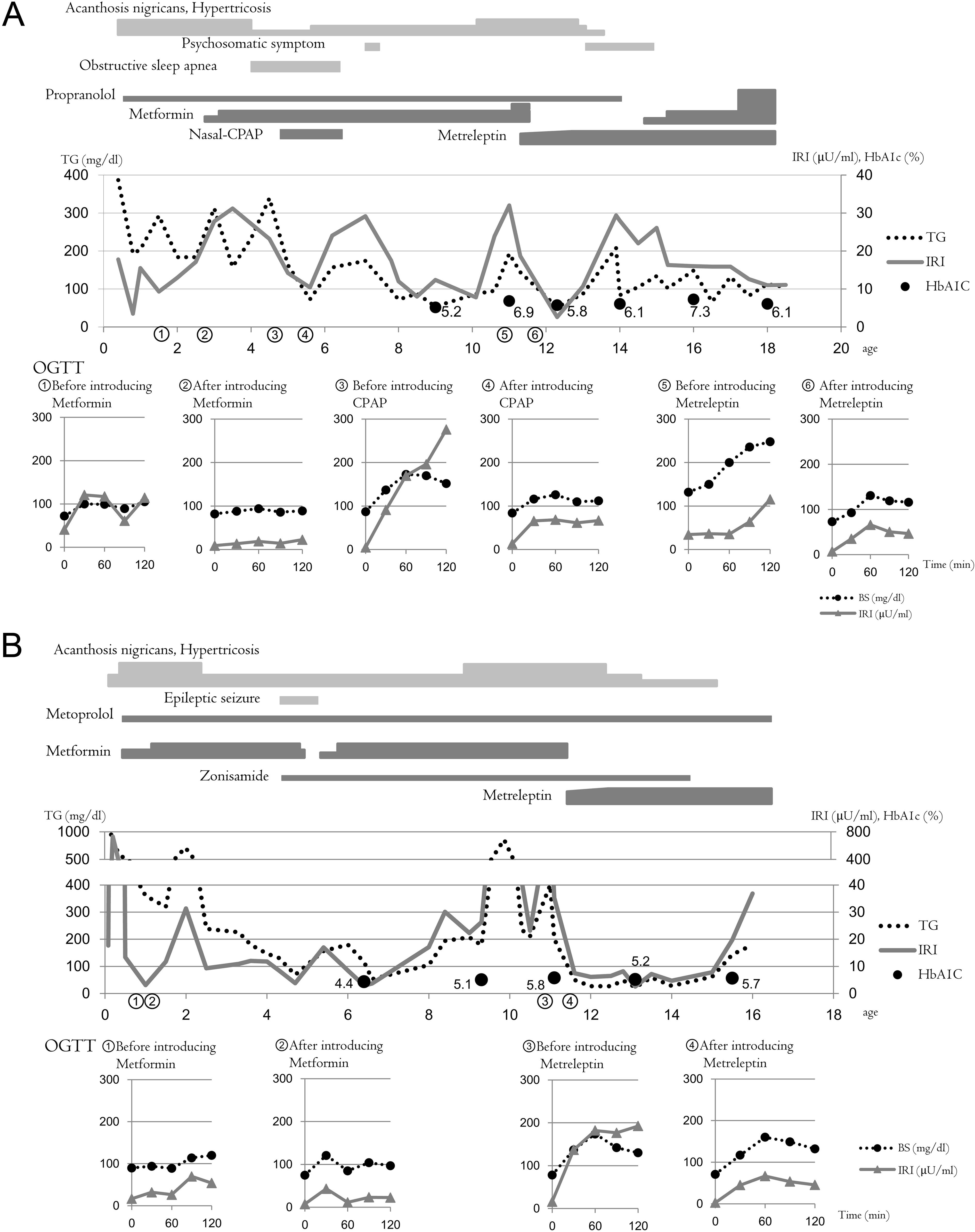 Download PDF (722K)
Download PDF (722K) -
2019 Volume 28 Issue 4 Pages 147-153
Published: 2019
Released on J-STAGE: October 19, 2019
 Download PDF (1898K)
Download PDF (1898K)
Mutation-in-Brief
-
2019 Volume 28 Issue 4 Pages 155-158
Published: 2019
Released on J-STAGE: October 19, 2019
 Download PDF (651K)
Download PDF (651K)
- |<
- <
- 1
- >
- >|