第34回反応と合成の進歩シンポジウム
選択された号の論文の179件中1~50を表示しています
2008年11月4日(火)
9:00~10:00 口頭発表 (座長 佐治木 弘尚)
-
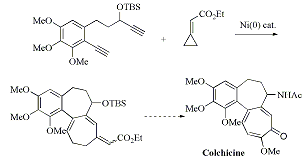
セッションID: 1O-1
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1O-2
発行日: 2008年
公開日: 2009/01/15
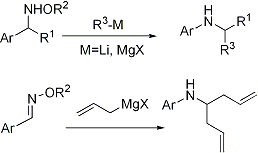
-
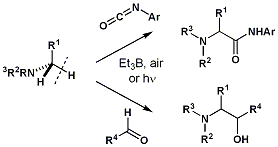
セッションID: 1O-3
発行日: 2008年
公開日: 2009/01/15
10:00~11:00 口頭発表 (座長 土井 隆行)
-
セッションID: 1O-4
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1O-5
発行日: 2008年
公開日: 2009/01/15
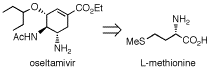
-
セッションID: 1O-6
発行日: 2008年
公開日: 2009/01/15

11:10~12:22 ポスターショートプレゼンテーション
-
セッションID: 1P-1
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-2
発行日: 2008年
公開日: 2009/01/15
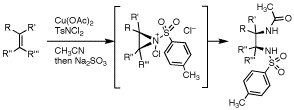
-
セッションID: 1P-3
発行日: 2008年
公開日: 2009/01/15
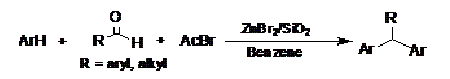
-
セッションID: 1P-4
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-5
発行日: 2008年
公開日: 2009/01/15
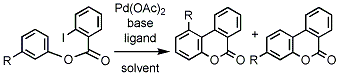
-
セッションID: 1P-6
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-7
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-8
発行日: 2008年
公開日: 2009/01/15
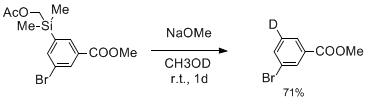
-
セッションID: 1P-9
発行日: 2008年
公開日: 2009/01/15
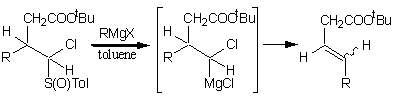
-
セッションID: 1P-10
発行日: 2008年
公開日: 2009/01/15
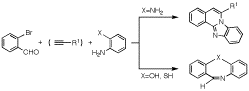
-
セッションID: 1P-11
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-12
発行日: 2008年
公開日: 2009/01/15
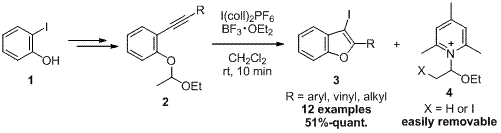
-
セッションID: 1P-13
発行日: 2008年
公開日: 2009/01/15
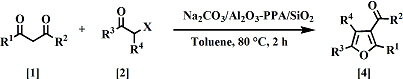
-
セッションID: 1P-14
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-15
発行日: 2008年
公開日: 2009/01/15
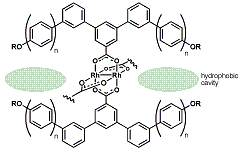
-
セッションID: 1P-16
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-17
発行日: 2008年
公開日: 2009/01/15
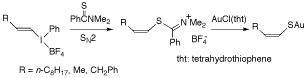
-
セッションID: 1P-18
発行日: 2008年
公開日: 2009/01/15
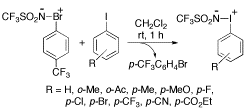
-
セッションID: 1P-19
発行日: 2008年
公開日: 2009/01/15
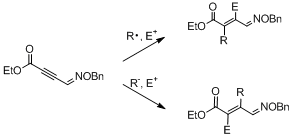
-
セッションID: 1P-20
発行日: 2008年
公開日: 2009/01/15
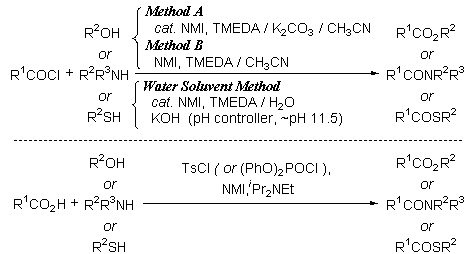
-
セッションID: 1P-21
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-22
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-23
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-24
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-25
発行日: 2008年
公開日: 2009/01/15
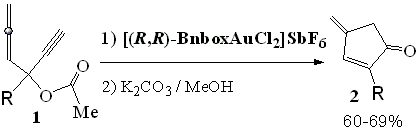
-
セッションID: 1P-26
発行日: 2008年
公開日: 2009/01/15
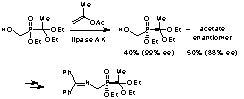
-
セッションID: 1P-27
発行日: 2008年
公開日: 2009/01/15
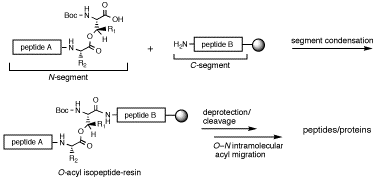
-
セッションID: 1P-28
発行日: 2008年
公開日: 2009/01/15
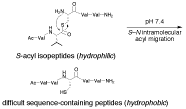
-
セッションID: 1P-29
発行日: 2008年
公開日: 2009/01/15
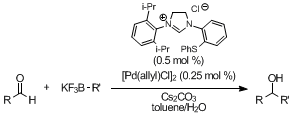
-
セッションID: 1P-30
発行日: 2008年
公開日: 2009/01/15
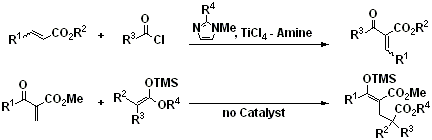
-
セッションID: 1P-31
発行日: 2008年
公開日: 2009/01/15
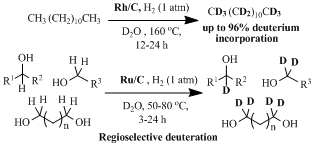
-
セッションID: 1P-32
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-33
発行日: 2008年
公開日: 2009/01/15
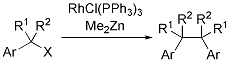
-
セッションID: 1P-34
発行日: 2008年
公開日: 2009/01/15
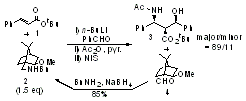
-
セッションID: 1P-35
発行日: 2008年
公開日: 2009/01/15
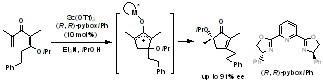
-
セッションID: 1P-36
発行日: 2008年
公開日: 2009/01/15
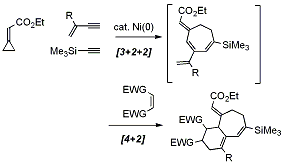
-
セッションID: 1P-37
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-38
発行日: 2008年
公開日: 2009/01/15
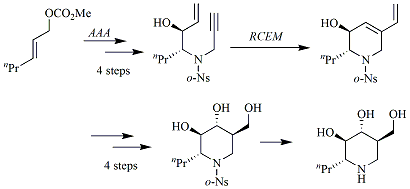
-
セッションID: 1P-39
発行日: 2008年
公開日: 2009/01/15
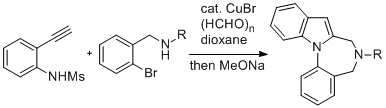
-
セッションID: 1P-40
発行日: 2008年
公開日: 2009/01/15

-
セッションID: 1P-41
発行日: 2008年
公開日: 2009/01/15
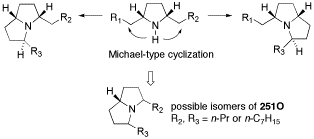
-
セッションID: 1P-42
発行日: 2008年
公開日: 2009/01/15
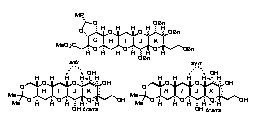
-
セッションID: 1P-43
発行日: 2008年
公開日: 2009/01/15
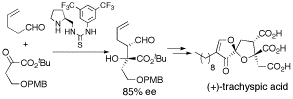
-
セッションID: 1P-44
発行日: 2008年
公開日: 2009/01/15