Abstract
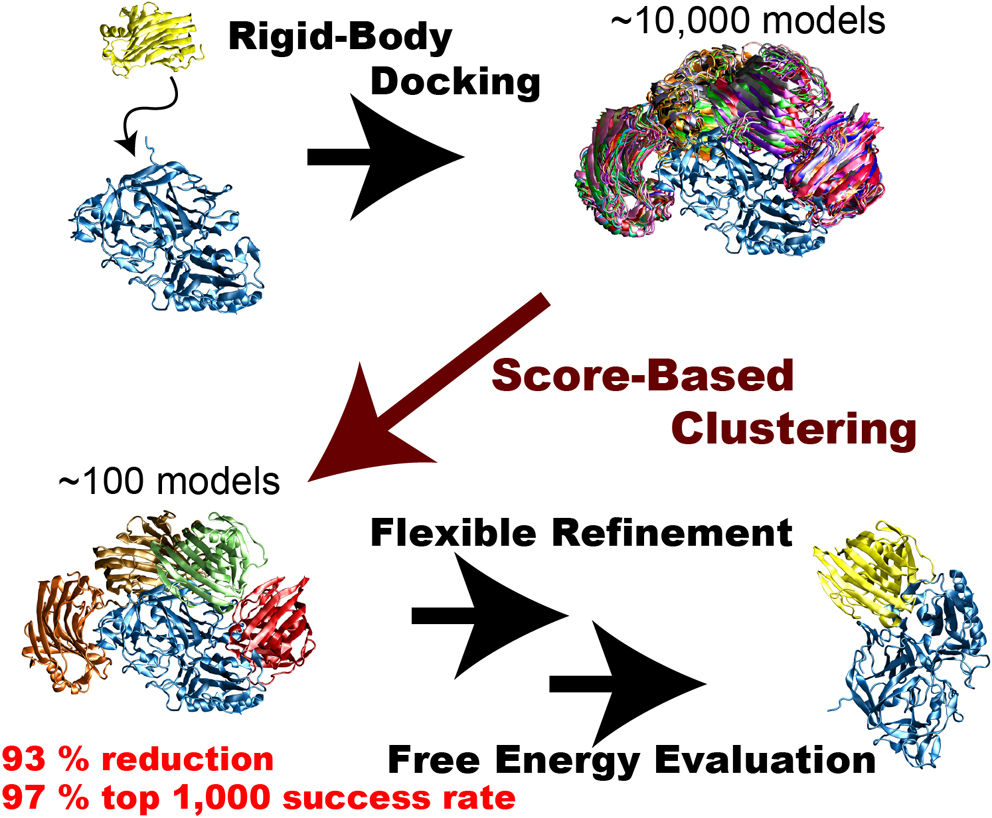
Rigid-body protein-protein docking is very efficient in generating tens of thousands of docked complex models (decoys) in a very short time without considering structure change upon binding, but typical docking scoring functions are not necessarily sufficiently accurate to narrow these decoys down to a small number of plausible candidates. Flexible refinements and sophisticated evaluation of the decoys are thus required to achieve more accurate prediction. Since this process is time-consuming, an efficient screening method to reduce the number of decoys is necessary immediately following rigid-body dockings. We attempted to develop an efficient screening method by clustering decoys generated by the rigid-body docking ZDOCK. We introduced the three metrics ligand-root-mean-square deviation (L-RMSD), interface-ligand-RMSD (iL-RMSD), and the fraction of common contacts (FCC), and examined various ranges of cut-offs for clusters to determine the best set of clustering parameters. Although the employed clustering algorithm is simple, it successfully reduced the number of decoys. Using iL-RMSD with a cut-off radius of 8 Å, the number of decoys that contain at least one near-native model with 90% probability decreased from 4,808 to 320, a 93% reduction in the original number of decoys. Using FCC for the clustering step, the top 1,000 success rates, defined as the probability that the top 1,000 models contain at least one near-native structure, reached 97%. We conclude that the proposed method is very efficient in selecting a small number of decoys that include near-native decoys.