Regular Article
-
Kentaro Ozawa, Hirotaka Taomori, Masayuki Hoshida, Ituki Kunita, Siger ...
2019Volume 16 Pages
1-8
Published: 2019
Released on J-STAGE: January 09, 2019
JOURNAL
FREE ACCESS
The movements of single actin filaments along a myosin-fixed glass surface were observed under a conventional fluorescence microscope. Although random at a low concentration, moving directions of filaments were aligned by the presence of over 1.0 mg/mL of unlabeled filaments. We found that actin filaments when at the intermediate concentrations ranging from 0.1 to 1.0 mg/mL, formed winding belt-like patterns and moved in a two-directional manner along the belts. These patterns were spread over a millimeter range and found to have bulged on the glass in a three-dimensional manner. Filaments did not get closer than about 37.5 nm to each other within each belt-pattern. The average width and the curvature radius of the pattern did not apparently change even when the range of actin concentrations was between 0.05 and 1.0 mg/mL or the sliding velocity between 1.2 and 3.2 μm/sec. However, when the length of filaments was shortened by ultrasonic treatments or the addition of gelsolin molecules, the curvature radius became small from 100 to 60 μm. These results indicate that this belt-forming nature of actin filaments may be due to some inter-filament interactions.
View full abstract
-
Hironobu Nogucci
2019Volume 16 Pages
9-17
Published: 2019
Released on J-STAGE: January 29, 2019
JOURNAL
FREE ACCESS
Supplementary material
The mechanical properties of tissues are influenced by those of constituent cells in various ways. For instance, it has been theoretically demonstrated that two-dimensional confluent tissues comprising mechanically uniform cells can undergo density-independent rigidity transitions, and analysis of the dynamical behavior of tissues near the critical point revealed that the transitions are geometrically controlled by the so-called cell shape parameter. To investigate whether three-dimensional tissues behave similarly to two-dimensional ones, we herein extend the previously developed model to three dimensions both dynamically and statically, demonstrating that two mechanical states similar to those of glassy materials exist in the three-dimensional case. Scaling analysis is applied to the static model focused from the rearrangement viewpoint. The obtained results suggest that the upper critical dimension of tissues equals two and is therefore the same as that of the jamming transition.
View full abstract
-
Shohei Konno, Kentaro Doi, Koichiro Ishimori
2019Volume 16 Pages
18-27
Published: 2019
Released on J-STAGE: January 31, 2019
JOURNAL
FREE ACCESS
Supplementary material
To investigate the dehydration associated with protein folding, the partial molar volume changes for protein unfolding (ΔVu) in cytochrome c (Cyt c) were determined using high pressure absorption spectroscopy. ΔVu values for the unfolding to urea- and guanidine hydrochloride (GdnHCl)-denatured Cyt c were estimated to be 56±5 and 29±1 mL mol–1, respectively. Considering that the volume change for hydration of hydrophobic groups is positive and that Cyt c has a covalently bonded heme, a positive ΔVu reflects the primary contribution of the hydration of heme. Because of the marked tendency of guanidium ions to interact with hydrophobic groups, a smaller number of water molecules were hydrated with hydrophobic groups in GdnHCl-denatured Cyt c than in urea-denatured Cyt c, resulting in the smaller positive ΔVu. On the other hand, urea is a relatively weak denaturant and urea-denatured Cyt c is not completely hydrated, which retains the partially folded structures. To unfold such partial structures, we introduced a mutation near the heme binding site, His26, to Gln, resulting in a negatively shifted ΔVu (4±2 mL mol–1) in urea-denatured Cyt c. The formation of the more solvated and less structured state in the urea-denatured mutant enhanced hydration to the hydrophilic groups in the unfolding process. Therefore, we confirmed the hydration of amino acid residues in the protein unfolding of Cyt c by estimating ΔVu, which allows us to discuss the hydrated structures in the denatured states of proteins.
View full abstract
-
Shuya Ishii, Madoka Suzuki, Shin’ichi Ishiwata, Masataka Kawai
2019Volume 16 Pages
28-40
Published: 2019
Released on J-STAGE: February 02, 2019
JOURNAL
FREE ACCESS
Supplementary material
The majority of hypertrophic cardiomyopathy (HCM) is caused by mutations in sarcomere proteins. We examined tropomyosin (Tpm)’s HCM mutants in humans, V95A and D175N, with in vitro motility assay using optical tweezers to evaluate the effects of the Tpm mutations on the actomyosin interaction at the single molecular level. Thin filaments were reconstituted using these Tpm mutants, and their sliding velocity and force were measured at varying Ca2+ concentrations. Our results indicate that the sliding velocity at pCa ≥8.0 was significantly increased in mutants, which is expected to cause a diastolic problem. The velocity that can be activated by Ca2+ decreased significantly in mutants causing a systolic problem. With sliding force, Ca2+ activatable force decreased in V95A and increased in D175N, which may cause a systolic problem. Our results further demonstrate that the duty ratio determined at the steady state of force generation in saturating [Ca2+] decreased in V95A and increased in D175N. The Ca2+ sensitivity and cooperativity were not significantly affected by the mutations. These results suggest that the two mutants modulate molecular processes of the actomyosin interaction differently, but to result in the same pathology known as HCM.
View full abstract
Review Article
Regular Article
-
Kenichi Kouyama, Kouta Mayanagi, Setsu Nakae, Yoshisuke Nishi, Masanao ...
2019Volume 16 Pages
59-67
Published: 2019
Released on J-STAGE: February 06, 2019
JOURNAL
FREE ACCESS
PolyADP-ribosylation (PARylation) is a posttranslational modification that is involved in the various cellular functions including DNA repair, genomic stability, and transcriptional regulation. PARylation is catalyzed by the poly(ADP-ribose) polymerase (PARP) family proteins, which mainly recognize damaged DNA and initiate repair processes. PARP inhibitors are expected to be novel anticancer drugs for breast and ovarian cancers having mutation in BRCA tumor suppressor genes. However the structure of intact (full-length) PARP is not yet known. We have produced and purified the full-length human PARP1 (h-PARP1), which is the major family member of PARPs, and analyzed it with single particle electron microscopy. The electron microscopic images and the reconstructed 3D density map revealed a dimeric configuration of the h-PARP1, in which two ring-shaped subunits are associated with two-fold symmetry. Although the PARP1 is hypothesized to form a dimer on damaged DNA, the quaternary structure of this protein is still controversial. The present result would provide the first structural evidence of the dimeric structure of PARP1.
View full abstract
-
Mika Sakamoto, Hirofumi Suzuki, Kei Yura
2019Volume 16 Pages
68-79
Published: 2019
Released on J-STAGE: February 15, 2019
JOURNAL
FREE ACCESS
Supplementary material
Transport of small molecules across the cell membrane is a crucial biological mechanism for the maintenance of the cell activity. ABC transporter family is a huge group in the transporter membrane proteins and actively transports the substrates using the energy derived from ATP hydrolysis. In humans, there are 48 distinct genes for ABC transporters. A variation of a single amino acid in the amino acid sequence of ABC transporter has been known to be linked with certain disease. The mechanism of the onset of the disease by the variation is, however, still unclear. Recent progress in the method to measure the structures of huge membrane proteins has enabled determination of the 3D structures of ABC transporters and the accumulation of coordinate data of ABC transporter has enabled us to obtain clues for the onset of the disease caused by a single variation of amino acid residue. We compared the structures of ABC transporter in apo and ATP-binding forms and found a possible conformation shift around pivot-like residues in the transmembrane domains. When this conformation change in ABC transporter and the location of pathogenic variation were compared, we found a reasonable match between the two, explaining the onset of the disease by the variation. They likely cause impairment of the pivot-like movement, weakening of ATP binding and weakening of membrane surface interactions. These findings will give a new interpretation of the variations on ABC transporter genes and pave a way to analyse the effect of variation on protein structure and function.

View full abstract
-
Yuhi Hosoe, Nobutaka Numoto, Satomi Inaba, Shuhei Ogawa, Hisayuki Mori ...
2019Volume 16 Pages
80-88
Published: 2019
Released on J-STAGE: February 22, 2019
JOURNAL
FREE ACCESS
Growth factor receptor-bound protein 2 (Grb2) is an adaptor protein that plays a critical role in cellular signal transduction. It contains a central Src homology 2 (SH2) domain flanked by two Src homology 3 (SH3) domains. Binding of Grb2 SH2 to the cytoplasmic region of CD28, phosphorylated Tyr (pY) containing the peptide motif pY-X-N-X, is required for costimulatory signaling in T cells. In this study, we purified the dimer and monomer forms of Grb2 SH2, respectively, and analyzed their structural and functional properties. Size exclusion chromatography analysis showed that both dimer and monomer exist as stable states. Thermal stability analysis using circular dichroism showed that the dimer mostly dissociates into the monomer around 50°C. CD28 binding experiments showed that the affinity of the dimer to the phosphopeptide was about three fold higher than that of the monomer, possibly due to the avidity effect. The present crystal structure analysis of Grb2 SH2 showed two forms; one is monomer at 1.15 Å resolution, which is currently the highest resolution analysis, and another is dimer at 2.00 Å resolution. In the dimer structure, the C-terminal region, comprising residues 123–152, was extended towards the adjacent molecule, in which Trp121 was the hinge residue. The stable dimer purified using size exclusion chromatography would be due to the C-terminal helix “swapping”. In cases where a mutation caused Trp121 to be replaced by Ser in Grb2 SH2, this protein still formed dimers, but lost the ability to bind CD28.
View full abstract
Review Article
-
Hiroshi Koyama, Dongbo Shi, Toshihiko Fujimori
2019Volume 16 Pages
89-107
Published: 2019
Released on J-STAGE: February 26, 2019
JOURNAL
FREE ACCESS
Organs and tissues in multi-cellular organisms exhibit various morphologies. Tubular organs have multi-scale morphological features which are closely related to their functions. Here we discuss morphogenesis and the mechanical functions of the vertebrate oviduct in the female reproductive tract, also known as the fallopian tube. The oviduct functions to convey eggs from the ovary to the uterus. In the luminal side of the oviduct, the epithelium forms multiple folds (or ridges) well-aligned along the longitudinal direction of the tube. In the epithelial cells, cilia are formed orienting toward the downstream of the oviduct. The cilia and the folds are supposed to be involved in egg transportation. Planar cell polarity (PCP) is developed in the epithelium, and the disruption of the Celsr1 gene, a PCP related-gene, causes randomization of both cilia and fold orientations, discontinuity of the tube, inefficient egg transportation, and infertility. In this review article, we briefly introduce various biophysical and biomechanical issues in the oviduct, including physical mechanisms of formation of PCP and organized cilia orientation, epithelial cell shape regulation, fold pattern formation generated by mechanical buckling, tubulogenesis, and egg transportation regulated by fluid flow. We also mention about possible roles of the oviducts in egg shape formation and embryogenesis, sinuous patterns of tubes, and fold and tube patterns observed in other tubular organs such as the gut, airways, etc.
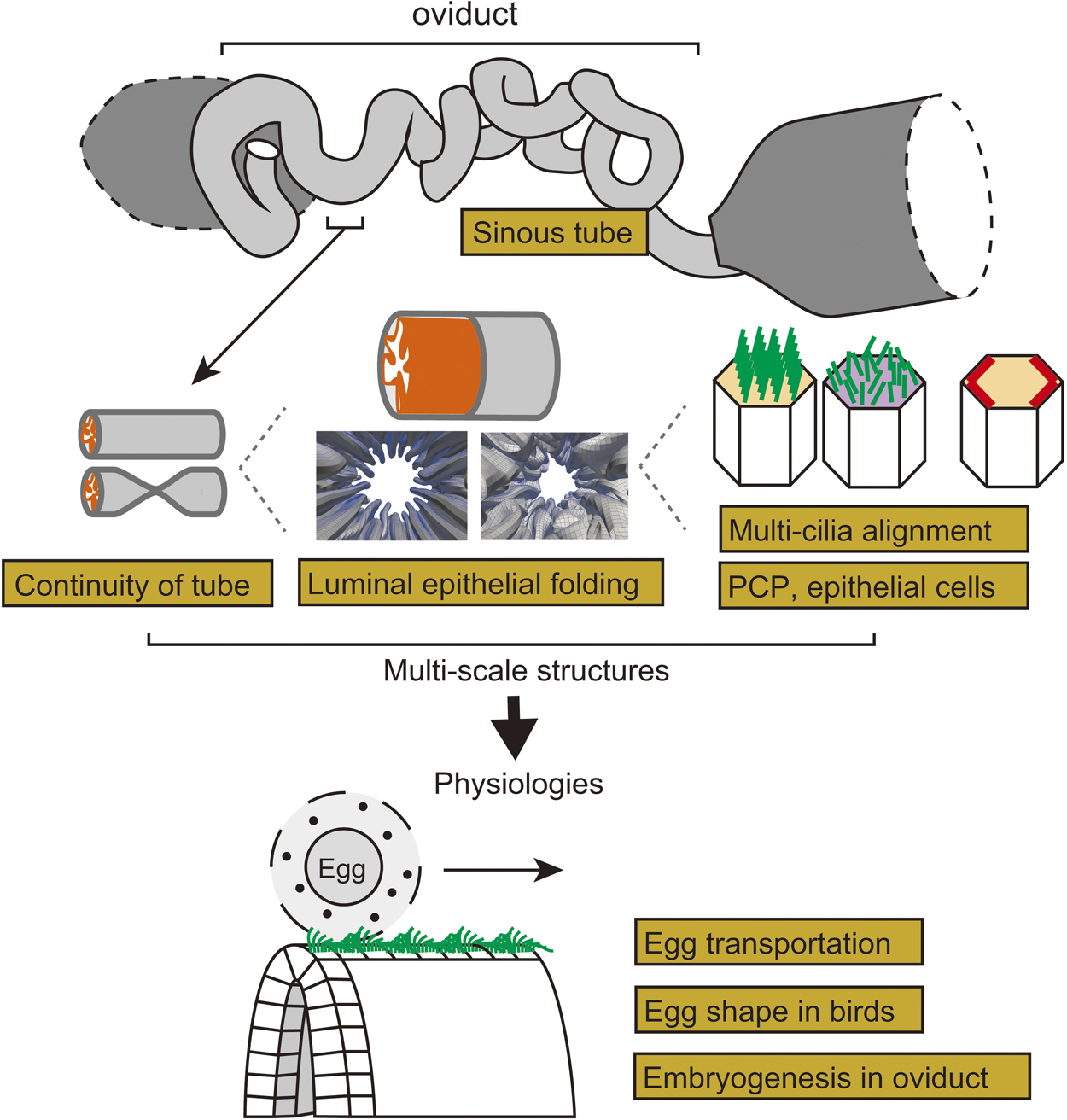
View full abstract
Regular Article
Review Article
-
Yosuke Tashiro, Kotaro Takaki, Hiroyuki Futamata
2019Volume 16 Pages
114-120
Published: 2019
Released on J-STAGE: April 19, 2019
JOURNAL
FREE ACCESS
Membrane vesicles (MVs) are lumen-containing spheres of lipid bilayers secreted by all prokaryotes into the extracellular milieu. They have multifunctional roles in stress response, virulence transfer, biofilm formation, and microbial interactions. Remarkably, MVs contain various components, including lytic enzymes, genetic materials, and hydrophobic signals, at high concentrations and transfer them effectively to the target microbial cells. Therefore, MVs act as carriers for bactericidal effects, horizontal gene transfer, and quorum sensing. Although the purpose of secreted MVs remains unclear, recent reports have provided evidence that MVs selectively interact with microbial cells in order to transfer their content to the target species. Herein, we review microbial interactions using MVs and discuss MV-mediated selective delivery of their content to target microbial cells.
View full abstract
-
Koichi Nakajo
2019Volume 16 Pages
121-126
Published: 2019
Released on J-STAGE: May 23, 2019
JOURNAL
FREE ACCESS
The KCNQ1 channel is a voltage-dependent potassium channel and is ubiquitously expressed throughout the human body including the heart, lung, kidney, pancreas, intestine and inner ear. Gating properties of the KCNQ1 channel are modulated by KCNE auxiliary subunits. For example, the KCNQ1-KCNE1 channel produces a slowly-activating potassium current, while KCNE3 makes KCNQ1 a voltage-independent, constitutively open channel. Thus, physiological functions of KCNQ1 channels are greatly dependent on the type of KCNE protein that is co-expressed in that organ. It has long been debated how the similar single transmembrane KCNE proteins produce quite different gating behaviors. Recent applications of voltage-clamp fluorometry (VCF) for the KCNQ1 channel have shed light on this question. The VCF is a quite sensitive method to detect structural changes of membrane proteins and is especially suitable for tracking the voltage sensor domains of voltage-gated ion channels. In this short review, I will introduce how the VCF technique can be applied to detect structural changes and what have been revealed by the recent VCF applications to the gating modulation of KCNQ1 channels by KCNE proteins.

View full abstract
Regular Article
-
Kei Odai, Tohru Sugimoto, Etsuro Ito
2019Volume 16 Pages
127-131
Published: 2019
Released on J-STAGE: August 03, 2019
JOURNAL
FREE ACCESS
5-Hydroxytryptamine (5-HT; serotonin) regulates metabolism and various homeostatic mechanisms in the body, and is involved in depression. These complicated functions of 5-HT are supported by several 5-HT receptor and 5-HT transporter subtypes. The development of agonists/antagonists and activators/inhibitors of 5-HT receptors and transporters is a strong target for drug studies. Toward this purpose, we calculated the conformations and electrical states of ionized 5-HT in aqueous solution using ab-initio methods. When we assumed an ionized 5-HT molecule and three surrounding water molecules, the hydrogen bond network for these four molecules formed a ring shape, resulting in deformation of the pyrrole ring in the indole group of 5-HT. To our knowledge, this is the first finding demonstrating deformation of the indole skeleton. The findings suggest that the direct involvement of water in the binding between 5-HT and its receptors and transporters should be taken account when designing candidate 5-HT active compounds.

View full abstract
Hypothesis and Perspective
-
Etsuro Ito, Rei Shima, Tohru Yoshioka
2019Volume 16 Pages
132-139
Published: 2019
Released on J-STAGE: August 24, 2019
JOURNAL
FREE ACCESS
We review the involvement of a small molecule, oxytocin, in various effects of physical stimulation of somatosensory organs, mindfulness meditation, emotion and fragrance on humans, and then propose a hypothesis that complex human states and behaviors, such as well-being, social bonding, and emotional behavior, are explained by oxytocin. We previously reported that oxytocin can induce pain relief and described the possibility how oxytocin in the dorsal horn and/or the dorsal root ganglion relieves joint and muscle pain. In the present article, we expand our research target from the physical analgesic effects of oxytocin to its psychologic effects to upregulate well-being and downregulate stress and anxiety. For this purpose, we propose a “hypothalamic-pituitary-adrenal (HPA) axis-oxytocin model” to explain why mindfulness meditation, placebo, and fragrance can reduce stress and anxiety, resulting in contentment. This new proposed model of HPA axis-oxytocin in the brain also provides a target to address other questions regarding emotional behaviors, learning and memory, and excess food intake leading to obesity, aimed at promoting a healthy life.
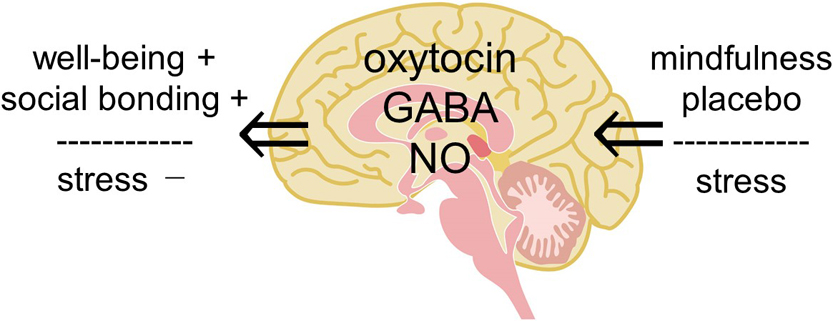
View full abstract
Review Article
-
Atsuko Nakanishi, Jun-ichi Kishikawa, Kaoru Mitsuoka, Ken Yokoyama
2019Volume 16 Pages
140-146
Published: 2019
Released on J-STAGE: September 03, 2019
JOURNAL
FREE ACCESS
Proton-translocating rotary ATPases couple proton influx across the membrane domain and ATP hydrolysis/synthesis in the soluble domain through rotation of the central rotor axis against the surrounding peripheral stator apparatus. It is a significant challenge to determine the structure of rotary ATPases due to their intrinsic conformational heterogeneity and instability. Recent progress of single particle analysis of protein complexes using cryogenic electron microscopy (cryo-EM) has enabled the determination of whole rotary ATPase structures and made it possible to classify different rotational states of the enzymes at a near atomic resolution. Three cryo-EM maps corresponding to different rotational states of the V/A type H+-rotary ATPase from a bacterium Thermus thermophilus provide insights into the rotation of the whole complex, which allow us to determine the movement of each subunit during rotation. In addition, this review describes methodological developments to determine higher resolution cryo-EM structures, such as specimen preparation, to improve the image contrast of membrane proteins.
View full abstract
Regular Article
-
Junichi Kaneshiro, Yasushi Okada, Tomohiro Shima, Mika Tsujii, Katsumi ...
2019Volume 16 Pages
147-157
Published: 2019
Released on J-STAGE: September 20, 2019
JOURNAL
FREE ACCESS
Supplementary material
Cryo-electron microscopy and X-ray crystallography have been the major tools of protein structure analysis for decades and will certainly continue to be essential in the future. Moreover, nuclear magnetic resonance or Förster resonance energy transfer can measure structural dynamics. Here, we propose to add optical second-harmonic generation (SHG), which is a nonlinear optical scattering process sensitive to molecular structures in illuminated materials, to the tool-kit of structural analysis methodologies. SHG can be expected to probe the structural changes of proteins in the physiological condition, and thus link protein structure and biological function. We demonstrate that a conformational change as well as its dynamics in protein macromolecular assemblies can be detected by means of SHG polarization measurement. To prove the capability of SHG polarization measurement with regard to protein structure analysis, we developed an SHG polarization microscope to analyze microtubules in solution. The difference in conformation between microtubules with different binding molecules was successfully observed as polarization dependence of SHG intensity. We also succeeded in capturing the temporal variation of structure in a photo-switchable protein crystal in both activation and inactivation processes. These results illustrate the potential of this method for protein structure analysis in physiological solutions at room temperature without any labeling.
View full abstract
-
Takeo Yamaguchi, Shunji Fukuzaki
2019Volume 16 Pages
158-166
Published: 2019
Released on J-STAGE: October 11, 2019
JOURNAL
FREE ACCESS
Supplementary material
Phosphorylation of membrane proteins in human erythrocytes is mediated by intracellular ATP levels. Such phosphorylation modulates the interactions of the bilayer with the cytoskeleton and affects the membrane stability under high pressure. In erythrocytes with high intracellular ATP levels, the bilayer-cytoskeleton interaction was weakened. Compression of such erythrocytes induced the release of large vesicles due to the suppression of fragmentation and resulted in the enhanced hemolysis. On the other hand, in ATP-depleted erythrocytes the interaction between the bilayer and the cytoskeleton was strengthened. Upon compression of these erythrocytes, the release of small vesicles due to the facilitation of vesiculation resulted in suppression of hemolysis. Taken together, these results suggest that the responses, i.e., vesiculation, fragmentation, and hemolysis, of the erythrocytes to high pressure are largely modulated by the bilayer-cytoskeleton interaction, which is mediated by intracellular ATP levels.
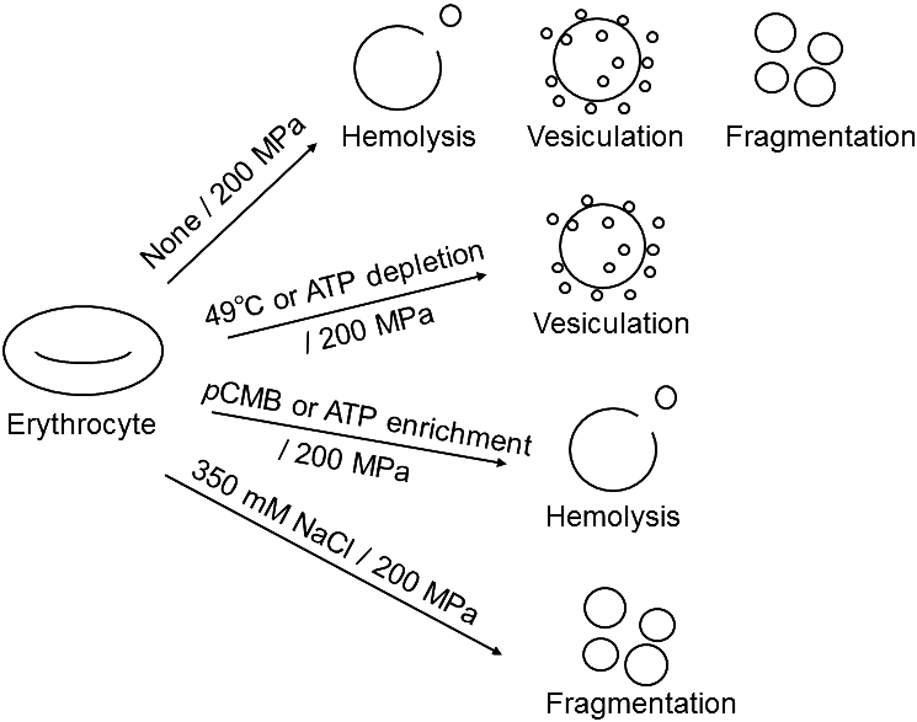
View full abstract
-
Kei Shimizu, Keita Ashida, Kohji Hotta, Kotaro Oka
2019Volume 16 Pages
167-172
Published: 2019
Released on J-STAGE: November 02, 2019
JOURNAL
FREE ACCESS
Exploring for food is important in food-deprived condition. Chemotaxis is one of the important behaviors to search food. Although chemotactic strategies in C. elegans have been well investigated: the pirouette and the weathervane strategies, the change of the chemotactic strategy by food deprivation is largely unclear. Here, we show the change of chemotactic strategy by food deprivation, especially for isoamyl alcohol. To compare chemotaxis under different food-deprivation period, we showed that worms change their chemotactic behaviors by food deprivation. The worms with 1-h food-deprivation change the weathervane strategy. On the other hand, 6-h food deprived animals change the pirouette strategy. These results demonstrate that worms change chemotactic strategy different way depend on period of food deprivation.
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Preface
-
Mikio Kataoka, Akio Kitao, Hidetoshi Kono
2019Volume 16 Pages
173-175
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Nobuyuki Matubayasi, Keiichi Masutani
2019Volume 16 Pages
185-195
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
The cosolvent effect on the equilibrium of peptide aggregation is reviewed from the energetic perspective. It is shown that the excess chemical potential is stationary against the variation of the distribution function for the configuration of a flexible solute species and that the derivative of the excess chemical potential with respect to the cosolvent concentration is determined by the corresponding derivative of the solvation free energy averaged over the solute configurations. The effect of a cosolvent at low concentrations on a chemical equilibrium can then be addressed in terms of the difference in the solvation free energy between pure-water solvent and the mixed solvent with the cosolvent, and illustrative analyses with all-atom model are presented for the aggregation of an 11-residue peptide by employing the energy-representation method to compute the solvation free energy. The solvation becomes more favorable with addition of the urea or DMSO cosolvent, and the extent of stabilization is smaller for larger aggregate. This implies that these cosolvents inhibit the formation of an aggregate, and the roles of such interaction components as the electrostatic, van der Waals, and excluded-volume are discussed.
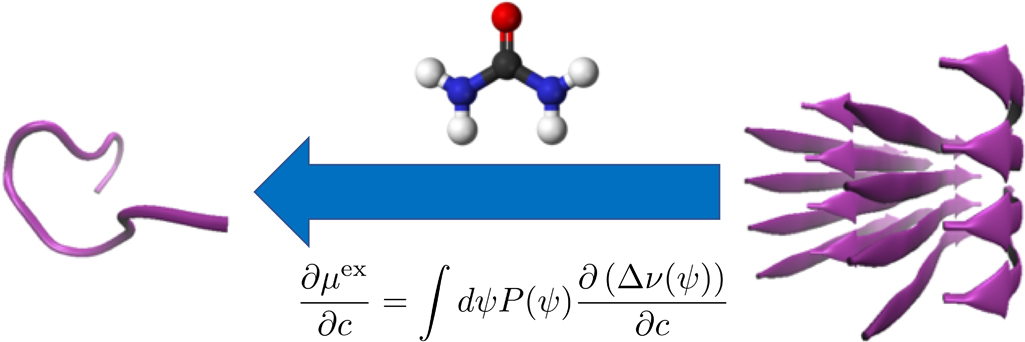
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Hiroshi Nakagawa, Mikio Kataoka
2019Volume 16 Pages
213-219
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Incoherent neutron scattering (INS) is one of the useful experimental methods for studying protein dynamics at the pico-nanosecond timescale. At this timescale, protein dynamics is highly coupled with hydration, which is observed as protein dynamical transition (PDT). INS is very sensitive to hydrogen atomic dynamics because of the large incoherent scattering cross section of hydrogen atom, and thus, the INS of a hydrated protein provides overall dynamic information about the protein, including hydration water. Separation of hydration water dynamics is essential for understanding hydration-related protein dynamics. H2O/D2O exchange is an effective method in the context of INS experiments for observing the dynamics of protein and hydration water separately. Neutron scattering is directly related to the van Hove space-time correlation function, which can be calculated quantitatively by performing molecular dynamics (MD) simulations. Diffusion and hydrogen bond dynamics of hydration water can be analyzed by performing MD simulation. MD simulation is useful for analyzing the dynamic coupling mechanism in hydration-related protein dynamics from the viewpoint of interpreting INS data because PDT is induced by hydration. In the present work, we demonstrate the methodological advantages of the H2O/D2O exchange technique in INS and the compatibility of INS and MD simulation as tools for studying protein dynamics and hydration water.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Shoji Takada
2019Volume 16 Pages
248-255
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
This review discusses Gō models broadly used in biomolecular simulations. I start with a brief description of the original lattice model study by Nobuhiro Gō. Then, the theory of protein folding behind Gō model, free energy approaches, and off-lattice Gō models are reviewed. I also mention a stringent test for the assumption in Gō models given from all-atom molecular dynamics simulations. Subsequently, I move to application of Gō models to protein dynamical functions. Various extension of Gō models is also reviewed. Finally, some publicly available tools to use Gō models are listed.
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Nobuhiro Go
2019Volume 16 Pages
256-263
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
The snake cube puzzle made of a linear array of 27 cubes and its modified and extended versions are used as theoretical models to study the mechanism of folding of proteins into their sequence-specific native three-dimensional structures. Each of the three versions is characterized by the respective set of characteristics attributed to each of its constituent cubes and an array is characterized by its specific sequence of the cube characteristics. The aim of the puzzles is to fold the cube array into a compact 3×3×3 cubic structure. In all three versions, out of all possible sequences, only a limited fraction of sequences are found foldable into the compact cube. Even among foldable sequences, the structures folded into the compact 3×3×3 cube are found often not uniquely determined from the sequence. By comparing the results obtained for the three versions of models, we conclude that the power of the hydrophobic interactions to make the folded structure unique to the sequence is much weaker than the geometrical varieties of constituent cubes as modelled in the original snake cube puzzle. However, when this weak cube attribute is compounded to that of the original snake cube puzzle, the power is enhanced very effectively. This is a strong manifestation of the consistency principle: The sequence-specific native structure of protein is realized as a result of consistency of various types of interactions working in protein.
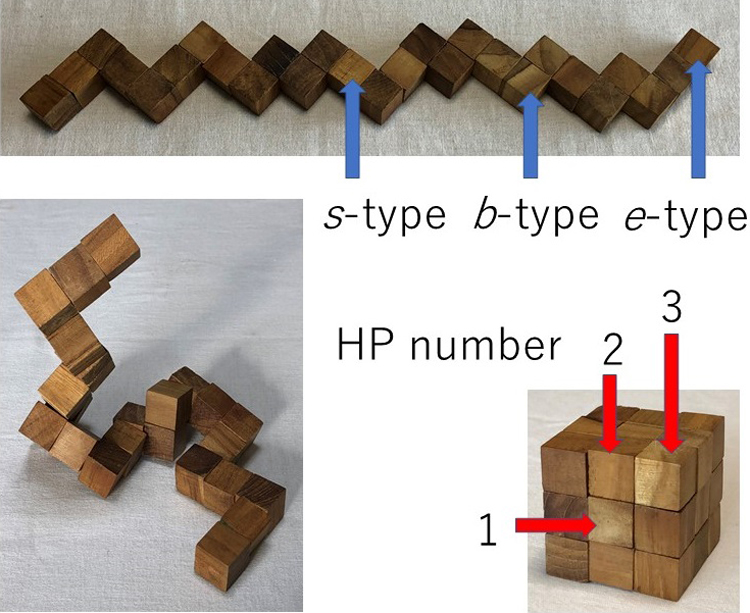
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Mikio Kataoka, Hironari Kamikubo
2019Volume 16 Pages
274-279
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
According to the consistency principle, a design principle for protein tertiary structures, all interactions that maintain a protein’s structure are consistent with each other. We assume that proteins satisfy the consistency principle. The specific local structures that form are consequences of the consistency principle. The specific local structures and the global conformation become interdependent. We assume that protein function is a consequence of the interdependency and the breaking of consistency. We applied this idea to the light-driven proton-pump mechanism of bacteriorhodopsin. Bacteriorhodopsin has two distinct conformers: one in which the proton channel opens toward the extracellular side, and another in which the channel opens toward the cytoplasmic side. Important reactions involved in proton pumping are protonation of D85 from the retinal Schiff base and reprotonation of the Schiff base from D96. To recruit a key water molecule, a characteristic pentameric hydrogen bond network is formed around the D85 and Schiff base, but is lost during proton pumping. These reaction components can be explained by active consistency-breaking and processes that either establish new consistency or restore the original consistency. Thus, the consistency principle can be expanded from structure to guide our understanding of protein function. This hypothesis is applicable to other functional proteins with two distinct conformers.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Kazuhiro Takemura, Akio Kitao
2019Volume 16 Pages
295-303
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Rigid-body protein-protein docking is very efficient in generating tens of thousands of docked complex models (decoys) in a very short time without considering structure change upon binding, but typical docking scoring functions are not necessarily sufficiently accurate to narrow these decoys down to a small number of plausible candidates. Flexible refinements and sophisticated evaluation of the decoys are thus required to achieve more accurate prediction. Since this process is time-consuming, an efficient screening method to reduce the number of decoys is necessary immediately following rigid-body dockings. We attempted to develop an efficient screening method by clustering decoys generated by the rigid-body docking ZDOCK. We introduced the three metrics ligand-root-mean-square deviation (L-RMSD), interface-ligand-RMSD (iL-RMSD), and the fraction of common contacts (FCC), and examined various ranges of cut-offs for clusters to determine the best set of clustering parameters. Although the employed clustering algorithm is simple, it successfully reduced the number of decoys. Using iL-RMSD with a cut-off radius of 8 Å, the number of decoys that contain at least one near-native model with 90% probability decreased from 4,808 to 320, a 93% reduction in the original number of decoys. Using FCC for the clustering step, the top 1,000 success rates, defined as the probability that the top 1,000 models contain at least one near-native structure, reached 97%. We conclude that the proposed method is very efficient in selecting a small number of decoys that include near-native decoys.
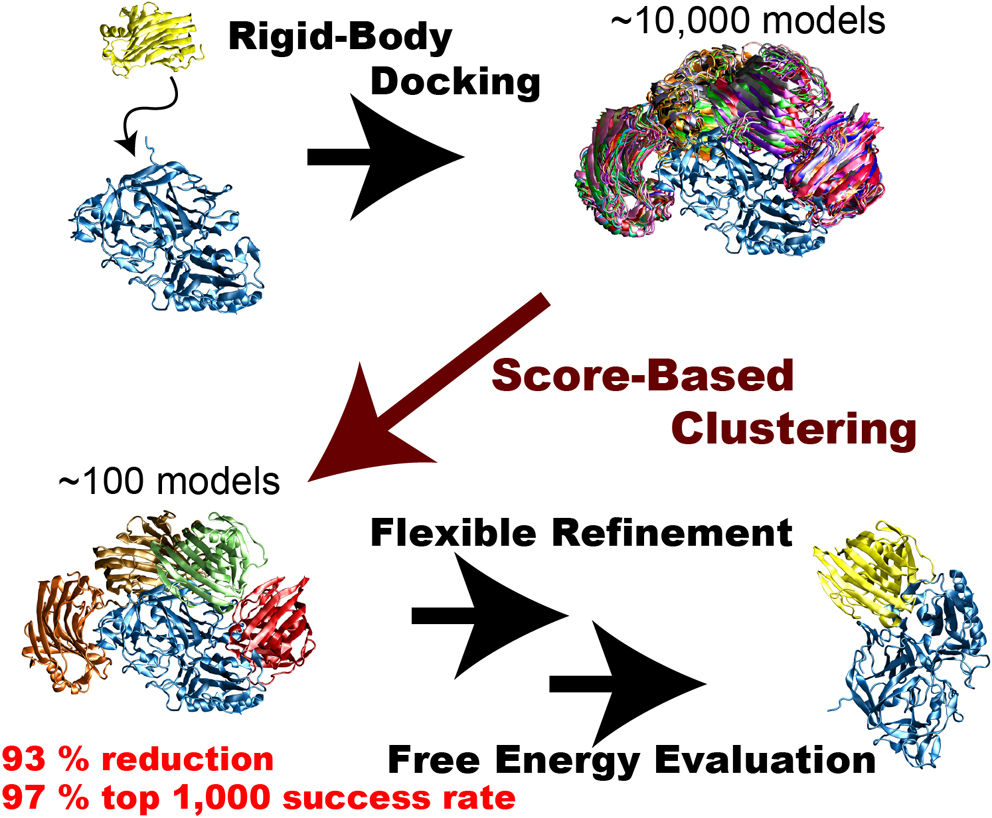
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Rie Koga, Nobuyasu Koga
2019Volume 16 Pages
304-309
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Protein design holds promise for applications such as the control of cells, therapeutics, new enzymes and protein-based materials. Recently, there has been progress in rational design of protein molecules, and a lot of attempts have been made to create proteins with functions of our interests. The key to the progress is the development of methods for controlling desired protein tertiary structures with atomic-level accuracy. A theory for protein folding, the consistency principle, proposed by Nobuhiro Go in 1983, was a compass for the development. Anfinsen hypothesized that proteins fold into the free energy minimum structures, but Go further considered that local and non-local interactions in the free energy minimum structures are consistent with each other. Guided by the principle, we proposed a set of rules for designing ideal protein structures stabilized by consistent local and non-local interactions. The rules made possible designs of amino acid sequences with funnel-shaped energy landscapes toward our desired target structures. So far, various protein structures have been created using the rules, which demonstrates significance of our rules as intended. In this review, we briefly describe how the consistency principle impacts on our efforts for developing the design technology.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Takahisa Yamato, Olivier Laprévote
2019Volume 16 Pages
322-327
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Normal mode analysis provides a powerful tool in biophysical computations. Particularly, we shed light on its application to protein properties because they directly lead to biological functions. As a result of normal mode analysis, the protein motion is represented as a linear combination of mutually independent normal mode vectors. It has been widely accepted that the large amplitude motions throughout the entire protein molecule can be well described with a few low-frequency normal modes. Furthermore, it is possible to represent the effect of external perturbations, e.g., ligand binding, hydrostatic pressure, as the shifts of normal mode variables. Making use of this advantage, we are able to explore mechanical properties of proteins such as Young’s modulus and compressibility. Within thermally fluctuating protein molecules under physiological conditions, tightly packed amino acid residues interact with each other through heat and energy exchanges. Since the structure and dynamics of protein molecules are highly anisotropic, the flow of energy and heat should also be anisotropic. Based on the harmonic approximation of the heat current operator, it is possible to analyze the communication map of a protein molecule. By using this method, the energy transfer pathways of photoactive yellow protein were calculated. It turned out that these pathways are similar to those obtained via the Green-Kubo formalism with equilibrium molecular dynamics simulations, indicating that normal mode analysis captures the intrinsic nature of the transport properties of proteins.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Ruth Veevers, Steven Hayward
2019Volume 16 Pages
328-336
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Domain movements play a prominent role in the function of many biomolecules such as the ribosome and F0F1-ATP synthase. As more structures of large biomolecules in different functional states become available as experimental techniques for structure determination advance, there is a need to develop methods to understand the conformational changes that occur. DynDom and DynDom3D were developed to analyse two structures of a biomolecule for domain movements. They both used an original method for domain recognition based on clustering of “rotation vectors”. Here we introduce significant improvements in both the methodology and implementation of a tool for the analysis of domain movements in large multimeric biomolecules. The main improvement is in the recognition of domains by using all six degrees of freedom required to describe the movement of a rigid body. This is achieved by way of Chasles’ theorem in which a rigid-body movement can be described as a screw movement about a unique axis. Thus clustering now includes, in addition to rotation vector data, screw-axis location data and axial climb data. This improves both the sensitivity of domain recognition and performance. A further improvement is the recognition and annotation of interdomain bending regions, something not done for multimeric biomolecules in DynDom3D. This is significant as it is these regions that collectively control the domain movement. The new stand-alone, platform-independent implementation, DynDom6D, can analyse biomolecules comprising protein, DNA and RNA, and employs an alignment method to automatically achieve the required equivalence of atoms in the two structures.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Yuko Okamoto
2019Volume 16 Pages
344-366
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
In this Special Festschrift Issue for the celebration of Professor Nobuhiro Gō’s 80th birthday, we review enhanced conformational sampling methods for protein structure predictions. We present several generalized-ensemble algorithms such as multicanonical algorithm, replica-exchange method, etc. and parallel Monte Carlo or molecular dynamics method with genetic crossover. Examples of the results of these methods applied to the predictions of protein tertiary structures are also presented.
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article
-
Yutaka Maruyama, Hiroshi Takano, Ayori Mitsutake
2019Volume 16 Pages
407-429
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Molecular dynamics simulation is a fruitful tool for investigating the structural stability, dynamics, and functions of biopolymers at an atomic level. In recent years, simulations can be performed on time scales of the order of milliseconds using specialpurpose systems. Since the most stable structure, as well as meta-stable structures and intermediate structures, is included in trajectories in long simulations, it is necessary to develop analysis methods for extracting them from trajectories of simulations. For these structures, methods for evaluating the stabilities, including the solvent effect, are also needed. We have developed relaxation mode analysis to investigate dynamics and kinetics of simulations based on statistical mechanics. We have also applied the three-dimensional reference interaction site model theory to investigate stabilities with solvent effects. In this paper, we review the results for designing amino-acid substitution of the 10-residue peptide, chignolin, to stabilize the misfolded structure using these developed analysis methods.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Atsushi Tokuhisa
2019Volume 16 Pages
430-443
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
An attainable structural resolution of single particle imaging is determined by the characteristics of X-ray diffraction intensity, which depend on the incident X-ray intensity density and molecule size. To estimate the attainable structural resolution even for molecules whose coordinates are unknown, this research aimed to clarify how these characteristics of X-ray diffraction intensity are determined from the structure of a molecule. The functional characteristics of X-ray diffraction intensity of a single biomolecule were theoretically and computationally evaluated. The wavenumber dependence of the average diffraction intensity on a sphere of constant wavenumber was observable by small-angle X-ray solution scattering. An excellent approximation was obtained, in which this quantity was expressed by an integral transform of the product of the external molecular shape and a universal function related to its atom packing. A standard model protein was defined by an analytical form of the first factor characterized by molecular volume and length. It estimated the numerically determined wavenumber dependence with a worst-case error of approximately a factor of five. The distribution of the diffraction intensity on a sphere of constant wavenumber was also examined. Finally, the correlation of diffraction intensities in the wavenumber space was assessed. This analysis enabled the estimation of an attainable structural resolution as a function of the incident X-ray intensity density and the volume and length of a target molecule, even in the absence of molecular coordinates.

View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Jean-François Gibrat
2019Volume 16 Pages
444-451
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
This paper presents a preliminary work consisting of two contributions. The first one is the design of a very efficient algorithm based on an “Overlap-Layout-Consensus” (OLC) graph to assemble the long reads provided by 3rd generation technologies. The second concerns the analysis of this graph using algebraic topology concepts to determine, in advance, whether the assembly of the genome will be straightforward, i.e., whether it will lead to a pseudo-Hamiltonian path or cycle, or whether the results will need to be scrutinized. In the latter case, it will be necessary to look for “loops” in the OLC assembly graph caused by unresolved repeated genomic regions, and then try to untie the “knots” created by these regions.
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
-
Nobuhiko Kajinami, Mitsuhiro Matsumoto
2019Volume 16 Pages
466-472
Published: 2019
Released on J-STAGE: November 29, 2019
JOURNAL
FREE ACCESS
Human knee joints move smoothly under high load conditions due to articular cartilage and synovial fluid. Much attention is paid to the role of proteoglycans. It is suggested that a part of proteoglycan forms aggregate on the cartilage surface, making a polymer brush, which has an important role in lubrication. In order to examine the lubrication mechanism in detail, we constructed a full atom model of a polymer brush system, and carried out a series of molecular dynamics simulations to analyze its frictional properties under constant shear. We use chondroitin 6-sulfate molecules grafted on resilient surface as the polymer brush and water with sodium ions as the synovial liquid. In the steady state, polymers have large deformation and the flow of synovial fluid becomes deviate from the Coutette flow, leading to a drastic reduction of friction. Longer chains have larger reduction.
View full abstract
Special Issue “Progress of Theoretical and Computational Biophysics”
Regular Article
Special Issue “Progress of Theoretical and Computational Biophysics”
Review Article

 Download PDF (3485K)
Download PDF (3485K) Download PDF (1970K)
Download PDF (1970K) Download PDF (6600K)
Download PDF (6600K) Download PDF (3537K)
Download PDF (3537K) Download PDF (1160K)
Download PDF (1160K) Download PDF (3377K)
Download PDF (3377K) Download PDF (4130K)
Download PDF (4130K) Download PDF (3502K)
Download PDF (3502K) Download PDF (1648K)
Download PDF (1648K) Download PDF (7219K)
Download PDF (7219K) Download PDF (2972K)
Download PDF (2972K) Download PDF (4464K)
Download PDF (4464K) Download PDF (6732K)
Download PDF (6732K)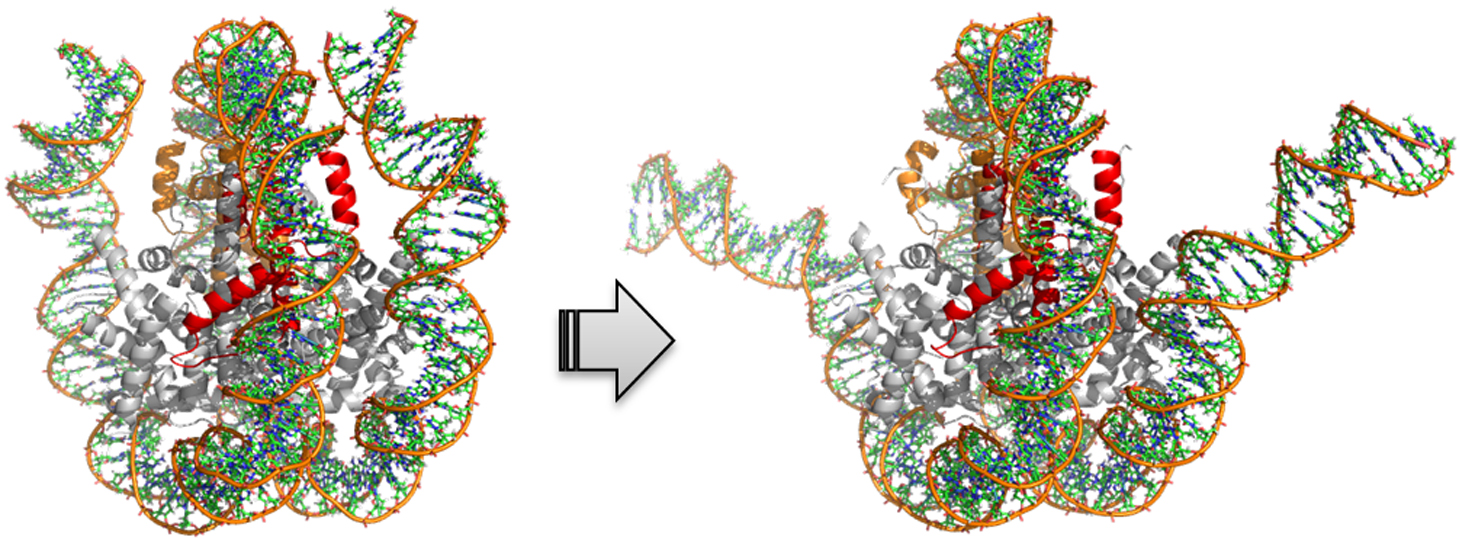 Download PDF (2369K)
Download PDF (2369K) Download PDF (8344K)
Download PDF (8344K)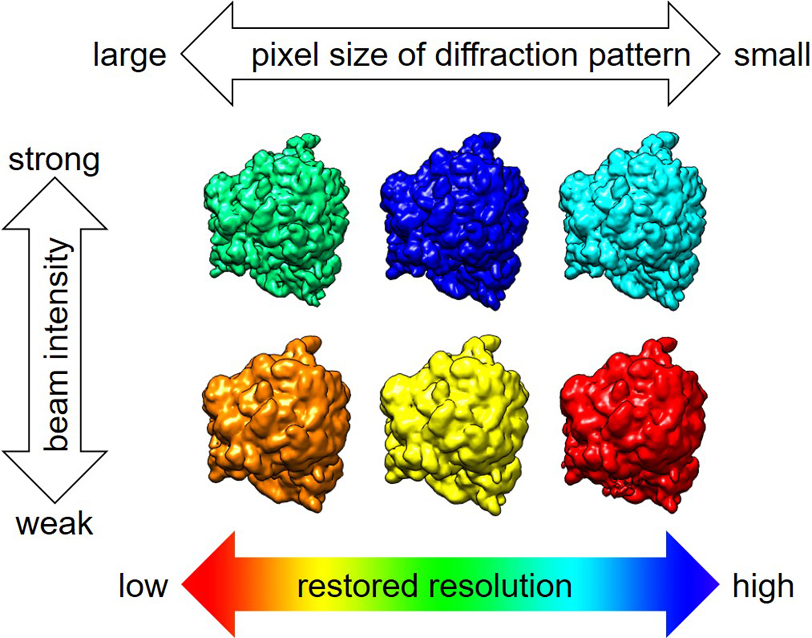 Download PDF (4622K)
Download PDF (4622K) Download PDF (5744K)
Download PDF (5744K) Download PDF (14863K)
Download PDF (14863K) Download PDF (1445K)
Download PDF (1445K) Download PDF (14918K)
Download PDF (14918K) Download PDF (5166K)
Download PDF (5166K) Download PDF (6751K)
Download PDF (6751K) Download PDF (1862K)
Download PDF (1862K)