Abstract
A transdermal oil-in-water type emulsified formulation containing propiverine hydrochloride, used for treatment of an overactive bladder (OAB), was evaluated for in vitro skin permeation under finite conditions and in vivo transdermal absorption. Propiverine hydrochloride solubility was determined using 1,3-butyleneglycol, polyoxyethylene (2) oleylether, isostearyl alcohol, and lauryl alcohol. The solubility increased as the solubility parameter value increased. In vitro skin permeation in hairless mouse skin and in vivo transdermal absorption in rats were measured using propiverine hydrochloride dissolved in a simple solution containing these solvents. Dependent on the increase in in vitro flux, the in vivo area under the curve up to 72 h (AUC0–72) was increased. Therefore, the emulsified formulation was prepared containing these ingredients using polyoxyethylene (20) stearylether for optimization. The emulsified formulation was used to conduct in vivo single- and repeated-dose absorption studies in rats. After single-dose transdermal administration of the emulsified formulation, the AUC0–72 was equivalent to that of the simple solution. Furthermore, results using the emulsified formulation indicated an increase in AUC0–72 and significant extension of the elimination half-life, in comparison with oral administration. After repeated-dose administration, a significant minimum plasma concentration was observed compared with oral administration. These results demonstrate that the emulsified formulation is a good option for transdermal delivery of propiverine hydrochloride.
Overactive bladder (OAB) is defined as a syndrome of urinary urgency accompanied by urinary frequency and nocturia.1) The symptoms of urinary incontinence is not required for OAB diagnosis; however, it occurs with increasing frequency with advancing age, in a similar manner to lower urinary tract symptoms.2) According to a recent epidemiological survey, 12% of Japanese adults, aged over 40 years, were estimated to be OAB sufferers.3) Although OAB is not a life-threatening disease, symptoms such as nocturia markedly impair QOL.4)
Propiverine hydrochloride, an anti-cholinergic drug, binds to muscarinic (M) receptors (mainly M3 receptors) of the detrusor and improves bladder storage function by suppressing involuntary muscle contraction.5) It has also been reported that propiverine relaxes the urinary bladder smooth muscle by a calcium antagonistic action. It is also suggested that it has an effect on atropine-resistant bladder contraction, in addition to the anti-cholinergic effects.6,7) Thus, propiverine has been used with oxybutynin, which is another anti-cholinergic drug, for the treatment of OAB for some time, because of its high efficacy and safety. In recent years, propiverine has been reported to improve stress urinary incontinence, a symptom unique to women, resulting in improved QOL for such female patients.8,9) In addition, propiverine reportedly has a more favorable tolerability profile compared with oxybutynin.6)
However, propiverine hydrochloride is currently available only as tablets or other oral dosage formulations, which have a poor adherence rate in OAB patients who hesitate to drink water, patients with kidney disease with water intake restriction, and patients with advanced age or dysphagia who are unable to smoothly swallow oral dosage formulations. To address these drawbacks, a transdermal formulation is explored as a new administration route that does not require water intake, thereby improving the treatment adherence of patients compared with that of oral administration.10)
Various techniques for preparing transdermal formulations have been developed to enhance permeation of numerous types of drugs through the stratum corneum and transdermal therapeutic systems have now been put to practical use to achieve not only local but also systemic effects. Oxybutynin is also used in a tape formulation.11) However, when continuous drug administration is required, such as in OAB treatment, tapes can have disadvantages such as causing skin irritation and necessitating application at different sites during repeated administration; thus, the dosage formulation discourages adherence. The possibility of transdermal absorption of propiverine has also been highlighted, but since no formulations with practically feasible administration conditions or a method for their evaluation have been established, the pharmacokinetics of propiverine following transdermal administration that are necessary for clinical evaluation, have not been elucidated.12) In recent years, a relationship between the active metabolite of propiverine and adverse reactions has been discussed. If transdermal administration can eliminate these adverse reactions by avoiding the first-pass effect, it could serve as a very interesting administration route.13)
In the present study, to develop a transdermal oil-in-water type emulsified formulation allowing sufficient transdermal absorption at a practically feasible dose, we conducted in vitro skin permeation and in vivo transdermal absorption studies using a finite dose under unocclusive conditions to establish the transdermal absorption of propiverine as a simple solution or an emulsified formulation.
MATERIALS AND METHODS
MaterialsPropiverine hydrochloride was purchased from Sanyo Chemical Laboratory Co., Ltd. (Toyama, Japan). 1,3-Butylene glycol (BG) was purchased from Daicel Corporation (Tokyo, Japan). Isostearyl alcohol (ISAL) was purchased from Kokyu Alcohol Kogyo Co., Ltd. (Chiba, Japan). Lauryl alcohol (LAL) and cetyl alcohol were purchased from New Japan Chemical Co., Ltd. (Osaka, Japan). Polyoxyethylene (2) oleylether (Oleth-2), polyoxyethylene (7) oleylether (Oleth-7), polyoxyethylene (10) oleylether (Oleth-10), polyoxyethylene (20) oleylether (Oleth-20), polyoxyethylene (50) oleylether (Oleth-50), polyoxyethylene (20) cetylether (Ceteth-20), polyoxyethylene (20) stearylether (Steareth-20), polyoxyethylene (20) behenylether (Beheneth-20), polyoxyl 40 stearate (MYS), and polyoxyethylene (20) polyoxypropylene (4) cetylether (PBC) were purchased from Nikko Chemicals Co., Ltd. (Tokyo, Japan). Dimethylpolysiloxane was purchased from Shin-Etsu Chemical Co., Ltd. (Niigata, Japan). Liquid paraffin was purchased from Shima Trading Co., Ltd. (Tokyo, Japan). Hydrogenated oil was purchased from Nippon Fine Chemical Co., Ltd. (Osaka, Japan). Sodium hydroxide (NaOH) was purchased from Kokusan Chemical Co., Ltd. (Tokyo, Japan). Sodium L-tartrate was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Methyl parahydroxybenzoate (MP) and ethyl parahydroxybenzoate (EP) were purchased from Ueno Fine Chemicals Industry, Ltd. (Osaka, Japan). All other chemicals used were of reagent grade.
Solubility Studies and Solubility Parameter CalculationTo establish the solubility values of propiverine hydrochloride in purified water, BG, Oleth-2, ISAL, and LAL, excess propiverine hydrochloride was added to each solvent and rotated with a micro tube rotor (As One, Osaka, Japan) at 30°C for 4 d. The solubility as propiverine was determined by HPLC after dilution with water or acetonitrile and filtrated with a 0.45 µm filter (Merck Millipore, Tokyo, Japan). Solubility parameters were calculated using Molecular Modelling Pro™ Plus software (ChemSW®, Fairfield, CA, U.S.A.) and values were calculated for the pure molecule of each ingredient based on the Van Krevelen and Hoftyzer approach.
Preparation of a Simple Solution Containing Propiverine HydrochlorideEach simple solution containing propiverine hydrochloride was prepared by weighing out the prescribed amount of each ingredient and stirring the mixture on a magnetic stirrer at room temperature. Table 1 shows the formulations (SOL1 to SOL6). All prepared simple solutions were transparent when the ingredients were completely dissolved.
Table 1. Composition of Simple Solutions Containing Propiverine Hydrochloride
| Ingredients | Formulation (%) |
|---|
| SOL1 | SOL2 | SOL3 | SOL4 | SOL5 | SOL6 |
|---|
| Propiverine hydrochloride | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| Sodium L-tartrate | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| BG | | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| Oleth-2 | | | 7.0 | 7.0 | 7.0 | 7.0 |
| ISAL | | | | 3.0 | | 3.0 |
| LAL | | | | | 3.0 | 3.0 |
| Purified water | 89.0 | 79.0 | 72.0 | 69.0 | 69.0 | 66.0 |
| Total | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
Table 2 shows each emulsified formulation (EF1 to EF10). Prescribed amounts of hydrogenated oil, dimethylpolysiloxane, LAL, cetyl alcohol, liquid paraffin, and ISAL were weighed out, mixed, and dissolved by warming to 70°C to obtain the oily phase (OP). Prescribed amounts of each surfactant (Oleth-2, -7, -10, -20, -50, Ceteth-20, Steareth-20, Beheneth-20, or MYS-40), PBC, BG, MP, and EP were weighed out, mixed, and dissolved by warming to 70°C, and then mixed with an appropriate volume of purified water at room temperature to obtain water phase 1 (WP1). Next, prescribed amounts of propiverine hydrochloride and sodium L-tartrate were weighed out and, after the addition of an appropriate volume of purified water, the mixture was dissolved by warming to 40°C to obtain water phase 2 (WP2). WP1 was mixed with OP by Homo Disper (PRIMIX Corporation, Osaka, Japan) to obtain a homogenous suspension, which was then mixed with WP2. In preparing EF14, NaOH was weighed out separately at a prescribed amount and dissolved in an appropriate amount of purified water at room temperature to obtain water phase 3 (WP3), which was then added to the homogenous suspension prepared by the Disper. A white emulsified formulation was obtained by leaving the final mixture at room temperature.
Table 2. Composition of Emulsified Formulations Containing Propiverine Hydrochloride
| Ingredients | Formulation (%) |
|---|
| EF1 | EF2 | EF3 | EF4 | EF5 | EF6 | EF7 | EF8 | EF9 | EF10 |
|---|
| Propiverine hydrochloride | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 |
| Sodium L-tartrate | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| BG | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 | 10.00 |
| Oleth-2 | 5.00 | | | | | | | | | |
| Oleth-7 | | 5.00 | | | | | | | | |
| Oleth-10 | | | 5.00 | | | | | | | |
| Oleth-20 | | | | 5.00 | | | | | | |
| Oleth-50 | | | | | 5.00 | | | | | |
| Ceteth-20 | | | | | | 5.00 | | | | |
| Steareth-20 | | | | | | | 5.00 | | | 7.00 |
| Beheneth-20 | | | | | | | | 5.00 | | |
| MYS | | | | | | | | | 5.00 | |
| MP | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 | 0.15 |
| EP | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 |
| PBC | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 |
| Hydrogenated oil | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 | 8.00 |
| Dimethylpolysiloxane | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 |
| Cetyl alcohol | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 7.00 |
| Liquid paraffin | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 2.00 |
| ISAL | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 |
| LAL | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 |
| NaOH | | | | | | | | | | 0.50 |
| Purified water | 47.25 | 47.25 | 47.25 | 47.25 | 47.25 | 47.25 | 47.25 | 47.25 | 47.25 | 45.75 |
| Total | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
The in vitro skin permeation of propiverine following delivery via the simple solutions or emulsified formulations was measured using Franz-type diffusion cells (effective diffusional area: 3.14 cm2, receptor volume: 17 mL). Laboskin® (excised dorsal skin of hairless mice, Hos: HR-1 male, 7 weeks) was purchased from Hoshino Laboratory Animals, Inc. (Saitama, Japan). Laboskin® was placed on the receptor cell with the stratum corneum facing upwards and in an unocclusive condition. Each simple solution or emulsified formulation was applied as a 5 mg formulation per cm2 of effective diffusional area of the stratum corneum surface, the donor chamber was clamped in place, and PBS (Dulbecco’s phosphate buffered saline, Sigma-Aldrich, St. Louis, MO, U.S.A.) added to the receptor cell. The receptor cell was stirred with a magnetic stirrer bar and maintained at 37°C in order to hold the temperature on the stratum corneum surface at 30°C. Aliquots (1.5 mL) were drawn from the receptor solution at 2, 4, 6, 8, 10, and 24 h and the same volume of fresh 37°C PBS was added to keep the volume constant. At the end of the experiment, any formulation remaining on the surface was removed using filter paper and the skin thickness measured by a micrometer (Mitutoyo Corporation, Kawasaki, Japan). The time-profiles of the cumulative permeated amounts of propiverine were calculated by HPLC determination of the propiverine concentrations in the collected receptor solutions.
In Vivo Absorption Study Following a Single-DoseSingle-dose administration was examined using a transdermal or oral administration. Male Sprague-Dawley rats aged 7–8 weeks (Japan SLC Inc., Shizuoka, Japan) were housed under a 12 h light/dark cycle in a room with controlled temperature (24±2°C) and humidity (55±5%). Food and water were provided ad libitum. To evaluate transdermal absorption, the dorsal skin of rats was sheared with clippers after anesthesia with pentobarbital (40 mg/kg, intraperitoneally (i.p.)). One hundred milligram of the simple solution or emulsified formulation was homogeneously applied to a 4×5 cm area of the dorsal section, kept in an unocclusive condition. To evaluate oral absorption, propiverine hydrochloride dissolved in water solution to 30 mg/kg/2 mL was administered to rats without anesthesia. Blood samples of 300 µL were collected from the subclavian vein without anesthesia at 1, 3, 6, 10, 24, 48, and 72 h after transdermal administration, or 0.5, 1, 3, 6, 10, 24, and 48 h in the case of oral administration. After each blood sample was centrifuged and filtered with a 0.45 µm filter, the plasma concentration of propiverine was measured by liquid chromatography tandem mass spectroscopy (LC-MS/MS). At the end of the collection period, the rat was euthanized by anesthesia with pentobarbital. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Shizuoka.
In Vivo Absorption Study Following Repeated-DosesMale Sprague-Dawley rats aged 7–8 weeks (Japan SLC Inc.) were housed under a 12 h light/dark cycle in a room with controlled temperature (24±2°C) and humidity (55±5%). Food and water were provided ad libitum. To evaluate transdermal absorption, the dorsal skin of rats was sheared with clippers after anesthesia with pentobarbital (40 mg/kg, i.p.). One hundred milligram of the emulsified formulation was homogeneously applied over a 4×5 cm area of the dorsal section, kept in an unocclusive condition. Blood samples of 300 µL were collected from the subclavian vein without anesthesia at 3, 6, 10, 24, 48, and 72 h after transdermal administration. After the 24 and 48 h blood sample collection, the skin application site was cleaned gently by cotton wool and 100 mg of the formulation re-administered without anesthesia. To evaluate oral absorption, propiverine hydrochloride dissolved in water solution to 43.5 mg/kg/2 mL was administered to rats without anesthesia. Blood samples of 300 µL were collected from the subclavian vein without anesthesia at 1, 3, 6, 10, 24, 48, and 72 h after oral administration. After the 24 and 48 h blood sample collection, propiverine hydrochloride dissolved in water solution to 43.5 mg/kg/2 mL was re-administered without anesthesia. Each blood sample was centrifuged and filtered with a 0.45 µm filter, the plasma concentration of propiverine was measured by LC-MS/MS. At the end of the collection period, the rat was euthanized by anesthesia with pentobarbital.
Determination of Propiverine Hydrochloride by in Vitro and in Vivo StudyThe receptor concentrations of propiverine in the in vitro study were determined by HPLC that consisted of a pump (LC-10ADvp, Shimadzu, Kyoto, Japan), an ultraviolet detector (SPD-10Avp, Shimadzu), a packed column (Capcell Pak® C18 MGIII, 3 µm, 3 mm i.d.×150 mm, Shiseido, Tokyo, Japan) and integrated by Labsolutions software (LCsolutions® v1.25, Shimadzu). The mobile phase was acetonitrile–distilled water (50 : 50, v/v) containing 0.1% phosphoric acid. Acetonitrile was used of HPLC grade (Wako Pure Chemical Industries, Ltd.). The flow rate was 0.5 mL/min. Detection was based on UV absorbance at 210 nm.
The plasma concentrations of propiverine in the in vivo study were determined by LC-MS/MS. LC-MS/MS was performed in an API4000 Q-TRAP® (Applied Biosystems, Foster City, CA, U.S.A.) equipped with HPLC (Prominence UFLC; Shimadzu). The separation was achieved on a Capcell Pak® C18 MGIII column (5 µm, 2 mm i.d.×150 mm, Shiseido) maintained at 40°C. A mobile phase consisting of solvent A (0.1% formic acid) and solvent B (acetonitrile) was delivered in a gradient from 65% A and 35% B over 3 min and then held to 10% A and 90% B over 6 min, then held for 1 min at a flow rate of 0.2 mL/min. Acetonitrile was used of LC-MS grade (Wako Pure Chemical Industries, Ltd.). The tandem mass spectrometer was operated at positive electrospray ionization (ESI) with a 4.5 kV ionization potential and an ion source temperature of 4500°C. The multiple reaction monitoring (MRM) transition ions were m/z 368.4/105.0 (Q1/Q3) for propiverine hydrochloride. Data acquisitions were ascertained by Analyst® 1.5 software (Applied Biosystems).
Data AnalysisThe apparent steady-state permeation flux (J0–10) in the in vitro permeation study was calculated using the cumulative amount of propiverine until 10 h and the least-squares method using the solver-function of Microsoft® Excel 2003. Pharmacokinetic parameters of the in vivo absorption study were calculated by a non-compartmental analysis method. The area under the plasma concentration versus time curve from 0 to 72 h (AUC0–72) was obtained using the trapezoidal rule. All data are presented as mean±standard deviation (S.D.). Statistical analysis was performed by Student’s t-test or Dunnett’s multiple comparison test using Graphpad Prism v.5.02 (Graphpad Software, San Diego, CA, U.S.A.). A p value of less than 0.05 was considered significant in all analyses.
RESULTS
Propiverine Solubility and Solubility Parameter ValuesSolubility values as propiverine in purified water, BG, Oleth-2, ISAL, and LAL are reported in Table 3. The solubility parameters of each ingredient are also given. Propiverine solubility increased as solubility parameter value increased.
Table 3. Solubility of Propiverine Hydrochloride at 30°C and Calculated Solubility Parameter Values (δ) of Each Solvent
| Solvents | Solubilitya) (mg/mL) | δb) (cal/cm3)1/2 |
|---|
| Purified water | 312±2.6 | 23.0 |
| BG | 7.8±0.4 | 14.1 |
| Oleth-2 | 1.9±0.06 | 9.45 |
| LAL | 0.72±0.02 | 9.19 |
| ISAL | 0.58±0.01 | 8.80 |
a) Each solubility value represents the mean±S.D. (n=3). b) All solubility parameters were calculated as pure molecules.
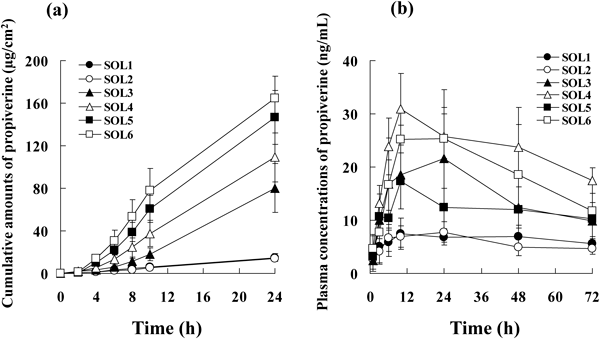
Figure 1(a) shows the time profiles for cumulative amounts of propiverine following administration as simple solutions SOL1 to SOL6 in the in vitro permeation study. Since the profiles showed a tendency to level off after 10 to 24 h, J0–10 was calculated from the gradient of the apparent steady-state profile up to 10 h. The J0–10 of SOL3, SOL4, and SOL5 were higher compared with SOL1 and SOL2, while SOL6 showed the highest value (Table 4). Figure 1(b) shows the time profile of plasma concentrations of propiverine from the in vivo single-dose transdermal absorption study. The calculated maximum plasma concentration (Cmax) and AUC0–72 showed that the AUC0–72 of SOL6 was significantly higher than those of SOL1, SOL2, and SOL5 (Table 4).
Table 4. Pharmacokinetic Parameters of Simple Solutions Containing Propiverine Hydrochloride from
in Vitro Permeation and
in Vivo Absorption Studies
| Formulation number | J0–10 (µg/cm2/h) | Cmax (ng/mL) | AUC0–72 (h·µg/mL) |
|---|
| SOL1 | 0.758±0.330 | 9.25±1.40 | 0.467±0.041 |
| SOL2 | 0.635±0.154 | 8.79±1.07 | 0.425±0.082 |
| SOL3 | 3.55±1.08 | 2.46±6.98*** | 1.08±0.269*** |
| SOL4 | 6.88±1.56** | 33.6±4.08*** | 1.67±0.168*** |
| SOL5 | 10.6±2.30*** | 17.5±2.91* | 0.870±0.088* |
| SOL6 | 12.5±2.30*** | 29.6±4.18*** | 1.38±0.273*** |
Each value represents the mean±S.D. (n=3–5). Asterisks show a significant difference in values compared with SOL1, using Dunnett’s multiple comparison test, * p<0.05, ** p<0.01, *** p<0.001.
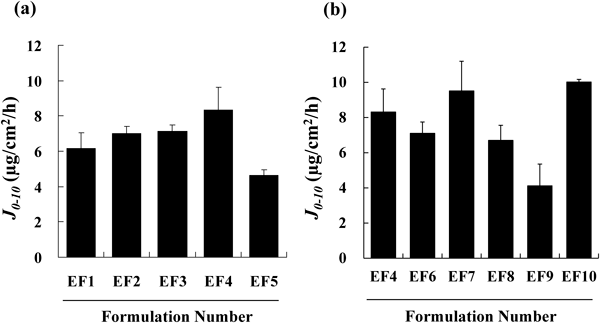
Emulsified formulations were prepared based on the composition of the simple solution, and the effects of adding various polyoxyethylene alkyl ethers as surfactants on the skin permeation of propiverine were compared. Figure 2(a) shows the J0–10 values obtained with the emulsified formulations containing Oleth-7, -10, -20, or -50 with different ethylene oxide chain lengths (7, 10, 20, 50 units of ethylene oxide (EO) per molecule, respectively), in comparison with the J0–10 obtained with Oleth-2, the surfactant that showed increased J0–10 with the simple solution (Table 4, EF1 to EF5). The J0–10 of the formulation containing Oleth-50 (EF5) tended to decrease relative to those of formulations containing the other Oleths. A similar comparison was made using polyoxyethylene alkyl ethers with different alkyl chain lengths, or MYS, a stearyl ester (Table 2, EF4 and EF6 to EF9). The J0–10 tended to decrease with MYS. Among the ethers, Steareth-20 showed the highest J0–10, which was 2.3-fold greater than that obtained with MYS (Fig. 2(b)). The J0–10 of EF10, which contains 7% Steareth-20 and is supplemented with NaOH, was 10.0±0.155 µg/cm2/h, which was not significantly different from the J0–10 of EF7 (9.52±1.69 µg/cm2/h).
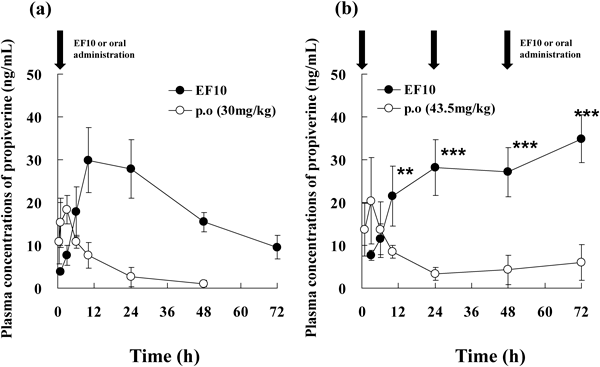
Evaluation of the in Vivo Absorption of Emulsified FormulationsThe in vivo transdermal absorption of propiverine from EF10 was compared with the absorption of orally administered propiverine hydrochloride delivered as a single-dose or repeated-doses. The propiverine hydrochloride dose was 43.5 mg/kg for all EF10 administrations, 30 mg/kg for single-dose oral administration, and 43.5 mg/kg for all repeated-dose administrations. Figures 3(a) and (b) show the plasma concentration profile of propiverine after single-dose and repeated-dose administration, respectively. Table 5 shows the pharmacokinetic parameters after single-dose oral administration. The AUC0–72 of EF10 was 5.7-fold higher than that for single-dose orally administered propiverine hydrochloride, which was a significant increase. The elimination half-life (t1/2) of EF10 was significantly longer than that observed after oral administration. With repeated-dose administration, the plasma concentration of propiverine after 72 h was 34.9±5.59 ng/mL for EF10, whereas the concentration following oral administration was only 5.97±4.21 ng/mL. Thus, the minimum plasma concentration of propiverine following the transdermal application of EF10 was 5.8 fold higher than that observed with repeated-dose oral administration, which was a significant increase.
Table 5. Pharmacokinetic Parameters from the
in Vivo Absorption Study after Administration of Oral and Transdermal Emulsified Formulations
| Parameter | Oral (30 mg/kg) | Transdermal (EF10) |
|---|
| t1/2 (h) | 16.3±8.06** | 36.4±15.0 |
| Tmax (h) | 2.43±0.980 | 14.0±6.83 |
| Cmax (ng/mL) | 19.0±4.01 | 30.9±7.38 |
| AUC0–72 (h·µg/mL) | 0.240±0.063* | 1.37±0.240 |
Each value represents the mean±S.D. (n=6–7). Asterisks show a significant difference from values with transdermal administration, using Dunnett’s multiple comparison test, * p<0.05, ** p<0.01.
DISCUSSION
For the development of a transdermal oil-in-water emulsified formulation of propiverine hydrochloride, basic in vitro and in vivo evaluations were performed on simple solutions under finite conditions. Based on the results obtained with the simple solutions, emulsified formulations were prepared and evaluated for transdermal absorption under finite conditions.
Evaluation of the simple solutions showed that propiverine solubility in the solvents used increased in a solubility parameter value-dependent manner (Table 3). Propiverine hydrochloride was completely dissolved in all simple solutions tested. However, in the in vitro skin permeation study performed under unocclusive conditions, water, the solvent with the highest propiverine solubility, evaporated during the course of the experiment. Therefore, it was assumed that the results shown in Table 3 allow estimation of the direct effect of each solvent on propiverine skin permeation in finite doses. Enhancement of J0–10 was not observed with SOL2, containing BG, whereas enhancement was observed with SOL3, containing Oleth-2, as well as with SOL4, 5, and 6, containing ISAL and (or) LAL. These non-ionic surfactants and long chain alcohols are well known to enhance skin permeation by increasing drug uptake into the stratum corneum.14,15) The results of the experiment with the simple solutions of propiverine in finite doses support the existence of an enhancing effect of these solvents. According to Higuchi’s equation, flux varies depending on the activity coefficient in the skin barrier when the activity of the drug applied to the skin is maximal, if the diffusivity and skin thickness are the same.16) Results of the propiverine solubility measurement (Table 3) suggest that, in the experiment performed under unocclusive conditions, the activity of propiverine increased because of water evaporation, even under finite conditions. Therefore, the results from Higuchi’s equation suggest that solubilized propiverine with Oleth-2, ISAL, and LAL decreased the activity coefficient of propiverine in the stratum corneum. These solvents interact easily with hydrophilic solvents from the reason of dissolution in preparing of simple solution. It is speculated that these solvent permeate into the skin with performing as co-solvent of water (and/or BG) and increase in the drug solubility in stratum corneum rather than individual component of Oleth-2, ISAL, and LAL. Thus, increased solubility in the stratum corneum appears to have contributed to the increase in J0–10.
In the in vitro skin permeation study, the cumulative propiverine concentration profile up to 10 h showed a flux under the apparent steady state, which leveled off because of the finite dose (Fig. 1(a)), precluding the judgment of whether or not the expected continuous flux could be achieved under in vivo finite conditions. To clarify this point, the in vivo transdermal absorption study was performed using simple solutions in finite doses. As a result, AUC0–72 increased in a manner dependent on in vitro J0–10, showing a significant increase when Oleth-2, ISAL, and LAL were added (Fig. 1(b)). On the other hand, ISAL increased both J0–10 and AUC, whereas LAL increased J0–10 alone (Table 4). In our present study, in vitro experiments were performed using hairless mouse dorsal skin, while in vivo experiments were performed using rats, the animal species commonly used in absorption studies. The usefulness of rats in the evaluation of transdermal absorption has previously been reported.17) ISAL and LAL, used in the present study, have relatively low propiverine hydrochloride solubility, with ISAL having a lower solubility parameter value than LAL (Table 3). Binary and ternary solvent systems acting as permeation enhancers are apt to be affected by the drug solubility and solubility parameters of individual solvents, providing a combination-dependent synergistic effect under infinite conditions.18) Given that ISAL has the lowest solubility parameter, the difference in the solubility parameter of the skin barrier of hairless mice compared with rats may have resulted in the difference in the uptake of propiverine.
The emulsified formulations were prepared based on the results obtained using simple solutions. In order to prepare stabilized emulsions, a greater number of different oily ingredients are required compared with the preparation of simple solutions, resulting in a complex system. Although it will be necessary to take into account the effects of the emulsified oily phase, which is very complex compared with a simple solution, in our present study, we investigated the effect of surfactant, the most critical ingredient in emulsified formulations, on in vitro flux. We found that the flux tended to decrease with Oleth-50, which has a relatively long EO chain, and with MYS, while no significant difference was observed with the other surfactants (Figs. 2(a), (b)). With an increase in EO chain length, the molecular weight of the surfactant increases accompanied by an increase in hydrophilicity, possibly decreasing the uptake of the surfactant on the stratum corneum. In order to further increase the stability of the emulsion, NaOH and an increased amount of Steareth-20 were added in the preparation of EF10 but these modifications had no effect on in vitro flux.
The level of transdermal absorption of propiverine with EF10 was compared with that following oral administration. The propiverine hydrochloride dose in EF10 was 43.5 mg/kg, which was slightly higher than the dose in oral administration (30 mg/kg), however, the plasma concentration changed more gradually after transdermal application than after oral administration, with significant increases in AUC0–72 and t1/2 observed with EF10. In this study, the absorption rate of propiverine was largely increased by preparing the emulsified formulation for the transdermal application. Thus, it was considered that the longer t1/2 observed in this study could be resulted in the increased absorption rate after transdermal administration. Furthermore, these results suggest that plasma propiverine concentration can be sufficiently maintained by in vivo transdermal emulsified formulations, even under unocclusive finite conditions. In the repeated-dose in vivo absorption study comparing oral administration and transdermal administration at the same propiverine hydrochloride dose (43.5 mg/kg), a higher minimum plasma propiverine concentration was maintained by transdermal administration than by oral administration over 72 h. These results suggest that the transdermal administration of propiverine hydrochloride under unocclusive finite conditions allows plasma propiverine concentration to be maintained at a high level without accumulation.
Thus, we have successfully validated an oil-in-water type emulsified formulation of propiverine hydrochloride with an excellent transdermal absorption profile.
Acknowledgments
The authors are grateful to Mr. Shinya Hirata, and Mr. Takumi Ikeda for their excellent technical assistance.
REFERENCES
- 1) Abrams P, Cardozo L, Fall M, Griffiths D, Rosier P, Ulmsten U, van Kerrebroeck P, Victor A, Wein A. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol. Urodyn., 21, 167–178 (2002).
- 2) Ouslander JG. Management of overactive bladder. N. Engl. J. Med., 350, 786–799 (2004).
- 3) Homma Y, Yamaguchi O, Hayashi K, Neurogenic Bladder Society Committee. An epidemiological survey of overactive bladder symptoms in Japan. BJU Int., 96, 1314–1318 (2005).
- 4) Takao T, Tsujimura A, Kiuchi H, Takezawa K, Okuda H, Yamamoto K, Fukuhara S, Miyagawa Y, Nonomura N. Correlation between overactive bladder symptoms and quality of life in Japanese male patients: focus on nocturia. Urology, 82, 189–193 (2013).
- 5) Resnick NM, Yalla SV, Laurino E. The pathophysiology of urinary incontinence among institutionalized elderly persons. N. Engl. J. Med., 320, 1–7 (1989).
- 6) Asimakopoulos AD, Cerruto MA, Del Popolo G, La Martina M, Artibani W, Carone R, Finazzi-Agrò E. An overview on mixed action drugs for the treatment of overactive bladder and detrusor overactivity. Urol. Int., 89, 259–269 (2012).
- 7) Yono M, Yoshida M, Wada Y, Kikukawa H, Takahashi W, Inadome A, Seshita H, Ueda S. Pharmacological effects of tolterodine on human isolated urinary bladder. Eur. J. Pharmacol., 368, 223–230 (1999).
- 8) Ushiroyama T, Ikeda A, Ueki M. Clinical efficacy of clenbuterol and propiverine in menopausal women with urinary incontinence: improvement in quality of life. J. Med., 31, 311–319 (2000).
- 9) Komatsu T, Gotoh M, Funahashi Y, Matsukawa Y, Sassa N, Kato K, Kato M, Hattori R. Efficacy of propiverine in improving symptoms and quality of life in female patients with wet overactive bladder. LUTS, 1, 20–24 (2009).
- 10) Schwantes U, Topfmeier P. Importance of pharmacological and physicochemical properties for tolerance of antimuscarinic drugs in the treatment of detrusor instability and detrusor hyperreflexia-chances for improvement of therapy. Int. J. Clin. Pharmacol. Ther., 37, 209–218 (1999).
- 11) Jirschele K, Sand PK. Oxybutynin: past, present, and future. Int. Urogynecol. J., 24, 595–604 (2013).
- 12) Ogiso T, Iwaki M, Hirota T, Tanino T, Muraoka O. Comparison of the in vitro skin penetration of propiverine with that of terodiline. Biol. Pharm. Bull., 18, 968–975 (1995).
- 13) Yamada S, Ito Y, Taki Y, Seki M, Nanri M, Yamashita F, Morishita K, Komoto I, Yoshida K. The N-oxide metabolite contributes to bladder selectivity resulting from oral propiverine: muscarinic receptor binding and pharmacokinetics. Drug Metab. Dispos., 38, 1314–1321 (2010).
- 14) Matsui R, Hasegawa M, Ishida M, Ebata T, Namiki N, Sugibayashi K. Skin permeation of lidocaine from crystal suspended oily formulations. Drug Dev. Ind. Pharm., 31, 729–738 (2005).
- 15) Lane ME. Skin penetration enhancers. Int. J. Pharm., 447, 12–21 (2013).
- 16) Higuchi T. Physical chemical analysis of percutaneous absorption process from creams and ointments. J. Soc. Cosmet. Chem., 11, 85–97 (1960).
- 17) Takeuchi H, Mano Y, Terasaka S, Sakurai T, Furuya A, Urano H, Sugibayashi K. Usefulness of rat skin as a substitute for human skin in the in vitro skin permeation study. Exp. Anim., 60, 373–384 (2011).
- 18) Hirata K, Helal F, Hadgraft J, Lane ME. Formulation of carbenoxolone for delivery to the skin. Int. J. Pharm., 448, 360–365 (2013).