Abstract
Alzheimer’s disease (AD) is a recognized incurable neurodegenerative disorder. Clinically prescribed medicines for AD are expected to bring about only slight symptomatic improvement or a delay of its progression. Another strategy, amyloid β (Aβ) lowing agents, has not been successful at memory improvement. We have hypothesized that an improvement in cognitive function requires the construction of neuronal networks, including neurite regeneration and synapse formation; therefore, we have been exploring candidates for radical anti-AD drugs that can restore Aβ-induced neurite atrophy and memory impairment. Our studies found several promising drug candidates that may improve memory dysfunction in AD model mice. The main activity of these drugs is the restoration of damaged axons. Focusing on candidates based on the recovery of neurite atrophy in vitro certainly leads to positive effects on memory improvement also in vivo. This suggests that neuronal network reconstruction may importantly relate to functional recovery in the brain. When identifying the signaling mechanisms of exogenous compounds like natural medicine-derived constituents, molecules directly activated by the compound are hard to be identified. However, the drug affinity responsive target stability (DARTS) analysis may pave the way to an approach to determine the initial molecule of the signaling pathway. Exploring new drug candidates and clarifying their signaling pathways directly relating to neuronal network reconstruction may provide promising therapeutic strategies with which to overcome AD.
1. INTRODUCTION
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disorder. Clinically prescribed medicines for AD can only provide slight symptomatic improvement or a delay of progress.1) These agents are insufficient for improving cognitive function in patients with more severe impairment and are associated with an increased risk of adverse events.2) Therefore, radical therapeutic drugs for AD need to be developed. Several strategies for lowering amyloid β (Aβ) have been investigated for decades in basic research and clinical trials as an alternative to symptomatic medicines. However, these approaches did not improve cognitive function in phase 2 and 3 trials.3,4)
We hypothesized that an improvement of cognitive function requires the construction of neuronal networks, including neurite regeneration and synapse formation; therefore, we have been exploring candidates for radical anti-AD drugs that can restore Aβ-induced neurite atrophy and improve memory dysfunction. Neuronal death is the end stage phenomenon in AD, but neuritic atrophy and synaptic loss begin at an earlier stage and directly cause memory deficits in AD.5,6) Even if neurites are damaged, surviving neurons can still extend or remodel neurites. We have focused on the reconstruction of neuronal networks, such as neurite regeneration and synaptic reformation as the essential event for the recovery of cognitive function after injury.7)
2. CRUDE DRUG EXTRACTS AND DERIVED COMPOUNDS TO REGENERATE Aβ-INDUCED AXONAL ATROPHY
Aβ is a major pathological cause of AD due to the formation of a β-sheet structure8); Aβ forms deposits in the brain, and subsequently induces neuronal cell death,9) neuritic atrophy,10,11) and synaptic loss.6) Aβ (25–35) is an active partial fragment of Aβ. This fragment also forms a β-sheet structure12) and damages neurons like full length peptide.12–17) We confirmed that Aβ25–35 induced axonal atrophy as potently as full-length Aβ1–42 in primary cultured cortical neurons.18)
3. WITHANOLIDE A, WITHANOSIDE IV AND WITHANOSIDE VI
Ashwagandha (roots of Withania somnifera DUNAL) is one of the most valuable herbal drugs used in Indian traditional medicine (Ayurveda) as a rasayana drug that is capable of imparting long life, youthful vigor, and good intellectual powers.19) Ashwagandha is clinically used for the treatment of general debility, nervous exhaustion, insomnia, loss of memory, and other conditions.20,21) These traditional uses imply that Ashwagandha may possibly be useful at improving neurodegenerative diseases. Indeed, this herbal drug has been reported to exert various pharmacological effects such as anti-inflammatory, anti-tumor, anti-oxidant, immunomodulatory, and anti-neuropsychiatric disease effects.22,23)
Withanolide A, withanoside IV, and withanoside VI were identified as active constituents that enhance neurite extension in human blastoma SH-SY5Y cells.24,25) These three compounds were individually administered to neurons displaying axonal atrophy, which later showed axonal regrowth.18,26,27) Rat cortical neurons formed synapses in vitro during 21-d culture. Thereafter, Aβ25–35 was administered to the neurons, resulting in decreased densities of presynapses and postsynapses.18) Nonetheless, post treatment with withanolide A, withanoside IV, or withanoside VI increased the synaptic densities.
These 3 compounds were also tested in vivo. Aβ25–35 was intracerebroventricularly (i.c.v.) injected into mice brains. Densities of axons and synapses in the parietal cortex were reduced, and spatial memory of the mice was diminished by the injection of Aβ25–35. Consecutive oral administration of withanolide A,18) withanoside IV,26) or withanoside VI27) for 12 d increased the densities of axons and synapses in the parietal cortex and improved spatial memory deficit.
Withanoside IV is a steroidal saponin conjugated with two glucose residues at position C3. After oral administration of withanoside IV, ‘sominone’ which is an aglycone of withanoside IV, but not withanoside IV itself, was detected as the main compound in the serum. This is due to metabolism by enterobacterial β-glucosidases.26) Sominone itself increased the densities of axons and dendrites and synaptic puncta in cultured cortical neurons under Aβ(25–35) treatment.28) These data suggest that sominone works at target organs by activating neuronal network formation after oral administration of withanoside IV.
4. METABOLITE 1
Ginseng, the roots of Panax ginseng, is widely used as a tonic medicine in traditional medicine. Significant improvement in learning and memory has been observed in brain-damaged rats29–31) and aged rats31) by oral administration of ginseng powder. In addition, the major ginseng saponins, ginsenosides Rb1 and Rg1, are known to enhance spatial learning in normal mice.32)
By our neurite outgrowth assay using human neuroblastoma SK-N-SH cells, protopanaxadiol (ppd)-type saponins such as ginsenosides Rb1 and Rb3 were active.33) Ginsenoside Rb1 as a typical ppd-type saponin in Ginseng Radix and its main metabolite M1 (20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol) were investigated for a memory improvement effect.33) In Aβ(25–35) i.c.v. injected AD model mice, ginsenoside Rb1 or M1 administration for 14 d significantly improved spatial memory deficits. When M1 was adminstered 3 d after Aβ(25–35) to cortical neurons, Aβ(25–35)-induced axonal atrophy was fully restored.34)
5. ASTRAGALOSIDES I, II AND IV ASTRAGALOSIDES I, II AND IV
Astragali Radix (roots of Astragalus mongholicus or A. membranaceus) is used mainly as a tonic agent in traditional Japanese Kampo medicine. A few reports had shown that Astragali Radix extract or its components activate neuronal function.
Our previous study found that administration of an aqueous extract of A. mongholicus improved memory deficit induced by Aβ(25–35) i.c.v. injection in mice.35) The extract-treated group showed increases in axonal densities and presynaptic densities in the cerebral cortex and hippocampus. The aqueous extract of A. mongholicus also enhanced axonal and presynaptic densities in Aβ(25–35)-treated cultured cortical neurons. Astragalosides I, II and IV, major components of Astragali Radix, also ameliorated Aβ(25–35)-induced axonal atrophy and presynaptic loss in the cortical neurons.
6. DIOSGENIN, ITS ACTIVITIES AND SIGNALING PATHWAY
As shown here, we found that the crude drug-derived steroidal sapogenins restored neurite atrophy and synaptic loss, resulting in memory improvement in AD model mice. Diosgenin, a major constituent in Dioscorea rhizome, is also a steroidal sapogenin. We focused on the possibility of diosgenin improving severe memory deficits in transgenic model mice of AD. Before our study, several therapeutic effects of diosgenin had been reported, including against cancer,36) food allergy,37) D-galactose-induced cognitive deficit38) and diabetic neuropathy.39) A diosgenin derivative, caprospinol, improves memory dysfunction in Aβ1–42-infused AD model rats via reduction of amyloid deposits.40)
5XFAD mice were used in our study. In the mouse, transgenes coding mutated human PS1 (M146L; L286V) and human APP (the Swedish mutations: K670N and M671L; the Florida mutation: I716V; and the London mutation: V717I) are co-overexpressed neuron-specifically.41) These five familial AD mutations increase levels of Aβ peptides in the brain, especially the neurotoxic peptide Aβ42. At 2 months of age, Aβ42 begins to be drastically increased and amyloid deposits are precipitated in the brain. Other AD transgenic mouse lines usually require a long period of more than 6–12 months for amyloid plaque formation and memory deficits.42) Memory deficits in 5XFAD mice were demonstrated after 4 months old by contextual fear conditioning43) and Y-maze test.41) We additionally confirmed impairment of spatial memory44) and object recognition memory45,46) in these mice.
In AD patients,5) 5XFAD mice47) and other AD model mice,48) axonal and synaptic degeneration was found. A diffused presynaptic staining co-localized with a bulb-like shaped swollen axon in the early and late stages of AD.49) The abnormal shaped axon terminal is supposed to be functionally degenerated. The core structure of degenerated axons and presynapses is called neuritic plaque, and is one of the pathological hallmarks of AD. That is, morphological degeneration of axons and presynapses indicates disruption of the neuronal network.
Object recognition memory performance in 5XFAD mice at age 6–8 months was significantly improved by diosgenin treatment.50) Amyloid plaques and neurofibrillary tangles in the cerebral cortex and hippocampus were significantly reduced by diosgenin administration.50) Interestingly, abnormally swollen patterns of axons and presynaptic terminals were observed in regions closely associated with amyloid plaques. These signs of degenerated axon terminals were reduced by diosgenin treatment (Fig. 1).
Aiming to identify a direct target protein for diosgenin in neurons, the drug affinity responsive target stability (DARTS) method51) was performed. DARTS is an established method to identify the direct binding proteins of small molecule compounds. The vulnerability of target proteins to the proteolysis reaction can be changed by binding to the compound. After proteolytic digestion, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or two dimensional (2D)-PAGE is performed to pick up thicker and thinner bands that appeared in the drug-treated sample, compared with the control sample. Protein identification is performed by MS analysis to obtain candidates for direct binding proteins.
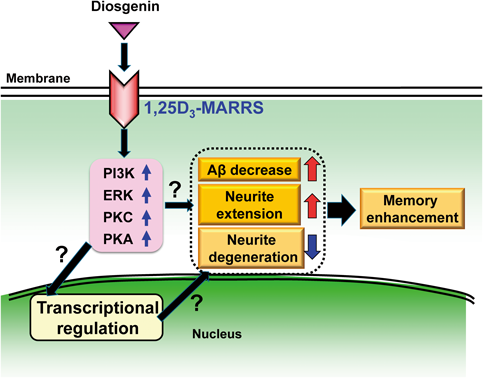
As a result, the membrane-associated rapid response steroid-binding receptor (1,25D3-MARRS) was proposed as a candidate of direct binding protein of diosgenin.50) This protein has multiple synonyms, which is dependent on subcellular locations.52,53) 1,25D3-MARRS is expressed on the cell surface and mediates the rapid response of 1α,25-dyhydroxyvitamin D3 (DHVD3).54) The rapid and non-genomic response of DHVD3 is mediated by the cell surface function of 1,25D3-MARRS.55) In contrast, the genomic effects of DHVD3 requires—binding to a nuclear receptor, nVDR, which activates gene expression through binding to DNA. 1,25D3-MARRS recruits multiple second messengers, at least phosphoinositide 3-kinase (PI3K), extracellular-response-activated kinase (ERK), protein kinase C (PKC), and protein kinase A (PKA).54,56,57)
Cell surface binding assay and in silico docking simulation provided evidence of direct binding of diosgenin to 1,25D3-MARRS.50) Functionally, 1,25D3-MARRS knockdown completely inhibited diosgenin-induced axonal growth in cortical neurons.50) The axonal regeneration effect of diosgenin in Aβ(1–42)-induced axonal atrophy was diminished by treatment with a neutralizing antibody for 1,25D3-MARRS.50)
In vivo conformation was also done using normal mice.58) Object recognition memory, spike firing and cross-correlation in the medial prefrontal cortex and hippocampal CA1 were facilitated by diosgenin administration. Axonal density and c-Fos expression were increased in the medial prefrontal and perirhinal cortices in the diosgenin-treated group, suggesting that neuronal excitation and morphological reinforcement may be enhanced. Under the sustained infusion of a neutralizing antibody for 1,25D3-MARRS, diosgenin treatment induced no memory enhancement or axonal growth. These in vivo data strongly suggest that diosgenin-elicited memory enhancement is mediated by 1,25D3-MARRS-triggered axonal growth.
To investigate which protein kinases are involved in diosgenin-induced axonal growth signaling, each specific inhibitor for PI3K, MEK1, PKC or PKA was co-applied with diosgenin. All inhibitors diminished diosgenin-induced axonal growth, suggesting that at least these kinases may be involved in the diosgenin signaling.50)
Our study was first to demonstrate that the exogenous stimulator diosgenin activates the 1,25D3-MARRS pathway. We are focusing on this signaling pathway as a very critical target for anti-AD therapy (Fig. 2).
7. USEFULNESS OF DARTS METHOD FOR IDENTIFYING UNKNOWN SIGNALING
In addition to single crude drug extract and pure compounds, we have investigated the anti-AD activities of traditional Japanese Kampo formulas, especially kamikihito (KKT). In Japanese Kampo medicine, KKT is thought to control insomnia, loss of appetite, amnesia, and depression. We reported improvement of memory dysfunction and axonal degeneration in 5XFAD mice by KKT.46) A small scale clinical study was performed using patients with AD and vascular dementia, and showed significant amelioration of memory function.59) Our study found that KKT treatment reversed Aβ-induced tau phosphorylation via protein phosphatase 2A (PP2A).60) However, inactivation of PP2A by Aβ and activation of PP2A by KKT in neurons was not robust. This suggested that other main signaling pathways might work in KKT-mediated axon regeneration. To confirm this possibility, the DARTS method was done to explore the primary target molecules of KKT in neurons. As a result, cytosolic aspartate aminotransferase (cAST) was identified as the most upstream molecule of KKT signaling.61) cAST knockdown or cAST inhibition diminished KKT-induced axonal growth.61) It is widely known that cAST converts L-aspartate and α-ketoglutarate to oxaloacetate and L-glutamate in the cytoplasm, and plays a crucial role in the malate–aspartate shuttle.62) However, cAST has never been thought to be included in the regulation of axonal regeneration.
DARTS methods may unravel the most upstream molecule of drug-driven signaling axis even if unpredictable signaling pathways would be worked.
8. CONCLUSION
Our studies found several drug candidates that may improve memory dysfunction in AD model mice (Table 1). The main activity of these drugs is the restoration of damaged axons. In our experience, the screening of natural drugs and traditional medicines results in dependable positive hits. Focusing on candidates based on the recovery of neurite atrophy in vitro certainly leads to positive effects on memory improvement in vivo also. This suggests that neuronal network reconstruction may be related to functional recovery in the brain. When identifying the signaling mechanism of exogenous compounds like natural medicine-derived constituents, molecules directly activated by the compound have not been identified easily. However, DARTS analysis may pave the way to an approach for identifying an initial molecule of the signaling pathway. Good drug seeds like diosgenin having many beneficial properties as anti-AD drugs would facilitate the establishment of new therapeutic strategies.
Table 1. Crude Drug Extracts and Identified Active Compounds That Restore Aβ-Induced Neurite Atrophy and Improve Memory Dysfunction
| Crude drug | Active compound | Restoration of Aβ-induced neurite atrophy | Memory improvement in AD model mice | References |
|---|
| Ashwagandha | Withanolide A | Axon, dendrite | Aβ(25–35) i.c.v. 5XFAD | 18), 25), 26), 27), 28), 45) |
| Withanoside IV | | | |
| Withanoside VI | | | |
| Sominone | | | |
| Panax ginseng | Ginsenoside Rb1 | Axon | Aβ(25–35) i.c.v. | 15), 33), 34) |
| Ginsenoside Rb3 | | | |
| M1 | | | |
| Astragalus mongholicus | Astragaloside I | Axon, dendrite | Aβ(25–35) i.c.v. | 35) |
| Astragaloside II | | | |
| Astragaloside IV | | | |
| Dioscorea batatas | Diosgenin | Axon, dendrite | 5XFAD | 50), 58) |
| Drynaria fortunei | (2S)-Neoeriocitrin caffeic acid 4-O-glucoside | Axon, dendrite | 5XFAD | 63) |
| Epimedium koreanum | Icariin | Axon, dendrite | 5XFAD | 44) |
| Eleutherococcus senticosus | Eleutheroside B | Axon, dendrite | Aβ(25–35) i.c.v. 5XFAD | 64), 65) |
| Eleutheroside E | | | |
| Isofraxidin | | | |
This review of the author’s work was written by the author upon receiving the 2016 Pharmaceutical Society of Japan Award for Divisional Scientific Promotion.
Acknowledgments
I would like to express my sincere gratitude to all collaborators described here. I also express cordial thanks to all members in my laboratory, both current and previous. This work was supported financially by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; Kampou Science Foundation; Uehara Memorial Foundation; Grant-in-Aid for the 21st Century COE Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Research Grant from the Astellas Foundation for Research on Metabolic Disorders; Grant-in-Aid for the Cooperative Research Project from Joint Usage/Research Center Institute of Natural Medicine, University of Toyama.
Conflict of Interest
The author declares no conflict of interest.
REFERENCES
- 1) Tan CC, Yu JT, Wang HF, Tan MS, Meng XF, Wang C, Jiang T, Zhu XC, Tan L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimer’s Dis., 41, 615–631 (2014).
- 2) Tricco AC, Soobiah C, Berliner S, Ho JM, Ng CH, Ashoor HM, Chen MH, Hemmelgarn B, Straus SE. Efficacy and safety of cognitive enhancers for patients with mild cognitive impairment: a systematic review and meta-analysis. Can. Med. Assoc. J., 185, 1393–1401 (2013).
- 3) Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M. Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer’s disease. Neurology, 73, 2061–2070 (2009).
- 4) Henley DB, May PC, Dean RA, Siemers ER. Siemers, E. R. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother., 10, 1657–1664 (2009).
- 5) Dickson TC, Vickers JC. The morphological phenotype of β-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience, 105, 99–107 (2001).
- 6) Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol., 30, 572–580 (1991).
- 7) Tohda C, Kuboyama T, Komatsu K. Search for natural products related to regeneration of the neuronal network. Neurosignals, 14, 34–45 (2005).
- 8) Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN, Li WY. Secondary structure of amyloid β peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol., 45, 373–379 (1994).
- 9) Bobinski M, Wegiel J, Tarnawski M, Bobinski M, Reisberg B, de Leon MJ, Miller DC, Wisniewski HM. Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer’s disease. J. Neuropathol. Exp. Neurol., 56, 414–420 (1997).
- 10) Canning DR, McKeon RJ, DeWitt DA, Perry G, Wujek JR, Frederickson RC, Silver J. Amyloid of Alzheimer’s disease induces reactive gliosis that inhibits axonal outgrowth. Exp. Neurol., 124, 289–298 (1993).
- 11) Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A., 96, 5274–5279 (1999).
- 12) Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure activity analyses of β-amyloid peptides: contribution of the β25–35 region to aggregation and neurotoxicity. J. Neurochem., 64, 253–265 (1995).
- 13) Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science, 250, 279–282 (1990).
- 14) Grace EA, Rabiner CA, Busciglio J. Characterization of neuronal dystrophy induced by fibrillar amyloid β: implications for Alzheimer’s disease. Neuroscience, 114, 265–273 (2002).
- 15) Sun MK, Alkon DL. Impairment of hippocampal CA1 heterosynaptic transformation and spatial memory by β-amyloid(25–35). J. Neurophysiol., 87, 2441–2449 (2002).
- 16) Tohda C, Tamura T, Komatsu K. Repair of amyloid β(25–35)-induced memory impairment and synaptic loss by a Kampo formula, Zokumei-to. Brain Res., 990, 141–147 (2003).
- 17) Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered β-amiloid peptides involves cholinergic dysfunction. Brain Res., 706, 181–193 (1996).
- 18) Kuboyama T, Tohda C, Komatsu K. Neuritic regeneration and synaptic reconstruction induced by withanolide A. Br. J. Pharmacol., 144, 961–971 (2005).
- 19) Sivarajan VV, Balachandran I. Ayurvedic drugs and their plant sources. International Science Publisher, New York (1994).
- 20) Warrier PK, Nambiar VPK, Ramankutty C. Indian Medicinal Plants: A Compendium of 500 Species. Orient Longman, Madras, India (1996).
- 21) Usmanghani K, Saeed A, Alam MT. Indusyunic medicine: traditional medicine of herbal, animal, and mineral origin in Pakistan. Dept. of Pharmacognosy, Faculty of Pharmacy, University of Karachi, Karachi, Pakistan (1997).
- 22) Kulkarni SK, Dhir A. Withania somnifera: an Indian ginseng. Prog. Neuropsychopharmacol. Biol. Psychiatry, 32, 1093–1105 (2008).
- 23) Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (Ashwagandha): a review. Altern. Med. Rev., 5, 334–346 (2000).
- 24) Zhao J, Nakamura N, Hattori M, Kuboyama T, Tohda C, Komatsu K. Withanolide derivatives from the roots of Withania somnifera and their neurite outgrowth activities. Chem. Pharm. Bull., 50, 760–765 (2002).
- 25) Kuboyama T, Tohda C, Zhao J, Nakamura N, Hattori M, Komatsu K. Axon- or dendrite-predominant outgrowth induced by constituents from Ashwagandha. Neuroreport, 13, 1715–1720 (2002).
- 26) Kuboyama T, Tohda C, Komatsu K. Withanoside IV and its active metabolite, sominone, attenuate Aβ(25–35)-induced neurodegeneration. Eur. J. Neurosci., 23, 1417–1426 (2006).
- 27) Tohda C, Komatsu K, Kuboyama T. Scientific basis for the anti-dementia drugs of constituents from Ashwagandha (Withania somnifera). J. Trad. Med., 22, 176–182 (2005).
- 28) Tohda C, Joyashiki E. Sominone enhances neurite outgrowth and spatial memory mediated by the neurotrophic factor receptor, RET. Br. J. Pharmacol., 157, 1427–1440 (2009).
- 29) Zhao R, McDaniel WF. Ginseng improves strategiclearning by normal and brain-damaged rats. Neuroreport, 9, 1619–1624 (1998).
- 30) Wang LC, Wang B, Ng SY, Lee TF. Effects of ginseng saponins on beta-amyloid-induced amnesia in rats. J. Ethnopharmacol., 103, 103–108 (2006).
- 31) Zhong YM, Nishijo H, Uwano T, Tamura R, Kawanishi K, Ono T. Red ginseng ameliorated place navigation deficits in young rats with hippocampal lesions and aged rats. Physiol. Behav., 69, 511–525 (2000).
- 32) Mook-Jung I, Hong HS, Boo JH, Lee KH, Yun SH, Cheong MY, Joo I, Huh K, Jung MW. Ginsenoside Rb1 and Rg1 improve spatial learning and increase hippocampal synaptophysin level in mice. J. Neurosci. Res., 63, 509–515 (2001).
- 33) Tohda C, Matsumoto N, Zou K, Meselhy RM, Komatsu K. Axonal and dendritic extension by protopanaxadiol-type saponins from Ginseng drugs in SK-N-SH cells. Jpn. J. Pharmacol., 90, 254–262 (2002).
- 34) Tohda C, Matsumoto N, Zou K, Meselhy MR, Komatsu KA. β(25–35)-induced memory impairment, axonal atrophy, and synaptic loss are ameliorated by M1, A metabolite of protopanaxadiol-type saponins. Neuropsychopharmacology, 29, 860–868 (2004).
- 35) Tohda C, Tamura T, Matsuyama S, Komatsu K. Promotion of axonal maturation and prevention of memory loss in mice by extracts of Astragalus mongholicus. Br. J. Pharmacol., 149, 532–541 (2006).
- 36) Yan LL, Zhang YJ, Gao WY, Man SL, Wang Y. In vitro and in vivo anticancer activity of steroid saponins of Paris polyphylla var. yunnanensis. Exp. Oncol., 31, 27–32 (2009).
- 37) Huang CH, Ku AY, Jan TR. Diosgenin attenuates allergen-induced intestinal inflammation and IgE production in a murine model of food allergy. Planta Med., 75, 1300–1305 (2009).
- 38) Chiu CS, Chiu YJ, Wu LY, Lu TC, Huang TH, Hsieh MT, Lu CY, Peng WH. Diosgenin ameliorates cognition deficit and attenuates oxidative damage in senescent mice induced by D-galactose. Am. J. Chin. Med., 39, 551–563 (2011).
- 39) Kang TH, Moon E, Hong BN, Choi SZ, Son M, Park JH, Kim SY. Diosgenin from Dioscorea nipponica ameliorates diabetic neuropathy by inducing nerve growth factor. Biol. Pharm. Bull., 34, 1493–1498 (2011).
- 40) Lecanu L, Rammouz G, McCourty A, Sidahmed EK, Greeson J, Papadopoulos V. Caprospinol reduces amyloid deposits and improves cognitive function in a rat model of Alzheimer’s disease. Neuroscience, 165, 427–435 (2010).
- 41) Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci., 26, 10129–10140 (2006).
- 42) Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav. Genet., 37, 79–100 (2007).
- 43) Ohno M. Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol. Learn. Mem., 92, 455–459 (2009).
- 44) Urano T, Tohda C. Icariin improves memory impairment in Alzheimer’s disease model mice (5xFAD) and attenuates amyloid β-induced neurite atrophy. Phytother. Res., 24, 1658–1663 (2010).
- 45) Joyashiki E, Matsuya Y, Tohda C. Sominone improves memory ipairments and increases axonal density in Alzheimer’s disease model mice, 5XFAD. Int. J. Neurosci., 121, 181–190 (2011).
- 46) Tohda C, Nakada R, Urano T, Okonogi A, Kuboyama T. Kamikihi-to (KKT) rescues axonal and synaptic degeneration associated with memory impairment in a mouse model of Alzheimer’s disease, 5XFAD. Int. J. Neurosci., 121, 641–648 (2011).
- 47) Zhang XM, Cai Y, Xiong K, Cai H, Luo XG, Feng JC, Clough RW, Struble RG, Patrylo PR, Yan XX. β-Secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur. J. Neurosci., 30, 2271–2283 (2009).
- 48) Trujillo-Estrada L, Dávila JC, Sánchez-Mejias E, Sánchez-Varo R, Gomez-Arboledas A, Vizuete M, Vitorica J, Gutiérrez A. Early neuronal loss and axonal/presynaptic damage is associated with accelerated amyloid-β accumulation in AßPP/PS1 Alzheimer’s disease mice subiculum. J. Alzheimer’s Dis., 42, 521–541 (2014).
- 49) Dickson TC, King CE, McCormack GH, Vickers JC. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp. Neurol., 156, 100–110 (1999).
- 50) Tohda C, Urano T, Umezaki M, Nemere I, Kuboyama T. Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer’s disease pathologies in 5XFAD mice. Sci. Rep, 2, 535 (2012).
- 51) Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U.S.A., 106, 21984–21989 (2009).
- 52) Wu W, Beilhartz G, Roy Y, Richard CL, Curtin M, Brown L, Cadieux D, Coppolino M, Farach-Carson MC, Nemere I, Meckling KA. Nuclear translocation of the 1,25D3-MARRS (membrane associated rapid response to steroids) receptor protein and NF-κB in differentiating NB4 leukemia cells. Exp. Cell Res., 316, 1101–1108 (2010).
- 53) Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD, Sehgal PB. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J. Interferon Cytokine Res., 22, 555–563 (2002).
- 54) Larsson B, Nemere I. Effect of growth and maturation on membrane-initiated actions of 1,25-dihydroxyvitamin D3-II: calcium transport, receptor kinetics, and signal transduction in intestine of female chickens. J. Cell. Biochem., 90, 901–913 (2003).
- 55) Nemere I, Farach-Carson MC, Rohe B, Sterling TM, Norman AW, Boyan BD, Safford SE. Ribozyme knockdown functionally links a 1,25(OH)2D3 membrane binding protein (1,25D3-MARRS) and phosphate uptake in intestinal cells. Proc. Natl. Acad. Sci. U.S.A., 101, 7392–7397 (2004).
- 56) Rosso A, Pansera M, Zamoner A, Zanatta L, Bouraïma-Lelong H, Carreau S, Silva FR. 1α,25(OH)2-Vitamin D3 stimulates rapid plasma membrane calcium influx via MAPK activation in immature rat Sertoli cells. Biochimie, 94, 146–154 (2012).
- 57) Buitrago C, Arango N, Boland R. 1α,25(OH)2D3-dependent modulation of Akt in proliferating and differentiating C2C12 skeletal muscle cells. J. Cell. Biochem., 113, 1170–1181 (2012).
- 58) Tohda C, Lee YA, Goto Y, Nemere I. Diosgenin-induced cognitive enhancement in normal mice is mediated by 1,25D3-MARRS. Sci. Rep, 3, 3395 (2013).
- 59) Magome A. Effect of kamikihito on dementia. Kampo Newest Ther., 23, 135–140 (2014).
- 60) Watari H, Shimada Y, Tohda C. New treatment for Alzheimer’s disease, kamikihito, reverses amyloid-β-induced progression of tau phosphorylation and axonal atrophy. Evid. Based Complement. Alternat. Med., 2014, 706487 (2014).
- 61) Watari H, Shimada Y, Tohda C. Cytosolic aspartate aminotransferase, a direct binding protein of kamikihito, regulates axon growth. Traditional Kampo Medicine, 3, 41–49 (2016).
- 62) McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem. Pharmacol., 71, 399–407 (2006).
- 63) Yang ZY, Kuboyama T, Kazuma K, Konno K, Tohda C. Active constituents from Drynaria fortunei Rhizomes on the attenuation of Aβ(25–35)-induced axonal atrophy. J. Nat. Prod., 78, 2297–2300 (2015).
- 64) Tohda C, Ichimura M, Bai Y, Tanaka K, Zhu S, Komatsu K. Inhibitory effects of Eleutherococcus senticosus extracts on amyloid β(25–35)-induced neuritic atrophy and synaptic loss. J. Pharmacol. Sci., 107, 329–339 (2008).
- 65) Bai Y, Tohda C, Zhu S, Hattori M, Komatsu K. Active components from Siberian ginseng (Eleutherococcus senticosus) for protection of amyloid β(25–35)-induced neuritic atrophy in cultured rat cortical neurons. J. Nat. Med., 65, 417–423 (2011).