Tuberculosis has been a well known infection throughout known history of humankind and was called “Captain Among These Men of Death” in 18th and 19th centuries. From 1882, the date when Mycobacterium tuberculosis was identified as the etiological agent of tuberculosis, till now scientists have struggled to discover efficient therapies against this contemporary infection.1) Approximately twenty years after WHO declared tuberculosis (TB) a “global public health emergency,” an estimated 8.6 million new case and 1.3 million death were noted in WHO’s Global Tuberculosis Report 2013.2) These large numbers of cases and deaths were based upon decreasing efficacy of four first line drugs; isoniazid, rifampicin, ethambutol, pyrazinamide and increasing resistance to at least isoniazid and rifampicin which was called multidrug-resistant (MDR) tuberculosis (TB) MDR-TB treatment comprises of second line antituberculosis drugs; ethionamide, prothionamide, thioacetazone, isoxyl (thiocarlide), amikacin, kanamycin or capreomycin as well as fluoroquinolone derivatives (ofloxacin, levofloxacin, moxifloxacin and gatifloxacin). Second line antituberculosis drugs are less potent, more toxic and more expensive than the first line drugs moreover anti-TB injectable drugs; amikacin, kanamycin or capreomycin decreases the therapy success rate due to their route of administration.3)
As a consequence of multiple mutations in specific resistant-associated genes of Mycobacterium tuberculosis (inhA, katG, rpoB, gyrA, rrs, tlyA and eis), extensively drug-resistant (XDR) TB has arisen. It was shown that treatment of XDR-TB by using isoniazid and rifampicin plus a fluoroquinolone derivative and amikacin, kanamycin or capreomycin is ineffectual.4) In view of the frequency and emergence of MDR and XDR tuberculosis and consequences of acquired resistance to clinically employed drugs, researchers have persisted in performing synthesis and anti-tuberculosis evaluation of novel compounds bearing various chemical entities.
Compounds with thiourea, acylthiourea and thioamide moiety have been synthesized as a result of drawing inspiration from second line antituberculosis pro-drugs; ethionamide (ETH), prothionamide, thiacetazone and isoxyl (thiocarlide).5–11) It was already known that, ETH inhibits cell wall biosynthesis of M. tuberculosis. Baulard et al. reported the identification of ETH-activator; Rv3854c which was then termed EthA.12) Another noteworthy work, revealed the mechanism of activation of ETH due to corresponding S-oxide by monooxygenase Rv3854c.13) It was noted that isoxyl activation also requires EthA-mediated oxidation.14) Isoxyl has been reported to be non-toxic in isoxyl-treated individulas at therapeutic doses but its poor solubility in water decreases its bioavalibility henceforth a new administration route-direct pulmonary delivery has been suggested due to refurbishment of old agents for emerging clinical needs.15) All of these phenomenon maintain undivided interest in thiourea synthesis. As well as thiourea based compounds, new candidates bearing both heterocycles and thiourea moieties have been shown as promising antituberculosis agents.7,8,10,16) It is known that heterocyclic scaffolds possess a leading role in designing novel class of chemotypes as drug candidates. Among them, 1,3,4-thiadiazoles have been reported to possess a wide range of biological activities including antiproliferative, antibacterial, antifungal, and antimycobacterial functions.17–21) Based on reported antitumor and uricogenic activity of 2-amino-1,3,4-thiadiazole (ATDA, NSC4728); Abdel Rahman and Mohamed22) synthesized 1,3-disubstituted thioureas via 5-(4-bromophenyl)-1,3,4-thiadiazol-2-amine. Some analogues from this series, that unite 1,3,4-thiadiazol ring with thiourea moiety, were reported to demonstrate promising IC50 values (2.58–6.47 µm) against A549 (Non-small Cell Lung Cancer) cell line.22) Another work on very similar chemotypes with our compounds has reported the synthesis of triazolothiadiazolethiones and thiadiazolothiadiazolimines that were gained by heterocyclization of N-(4-chlorophenyl)/phenyl-N′-[5-(4-methoxyphenyl)/(2-chlorophenyl)/phenyl-1,3,4-thiadiazol-2-yl]thioureas and N-phenyl/(2-chlorophenyl)-N′-(5-phenyl-1,3,4-thiadiazol-2-yl)thioureas as well as their antifungal activity against Aspergillus niger and Fusarium oxysporium.23)
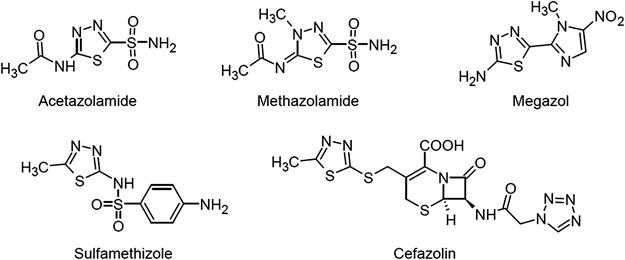
Actually, 1,3,4-thiadiazole ring is found in several drugs in clinical use. Typical examples are Acetazolamide and Methazolamide (carbonic anhydrase inhibitors); Sulfamethizole and Cefazolin (antibacterials) and Megazol (treatment of human African trypanosomiasis) (Fig. 1). We have recently reported two groups of novel 1,3,4-thiadiazole–4-thiazolidinone hybrids which were active against hepatitis C virus (HCV) via inhibition of NS5B polymerase.24,25) It is also worth to mention that M. tuberculosis contains three β-carbonic anhydrase (CA) genes in its genome; Rv1284, Rv3588c and Rv3273 encoding for mtCA 1, mtCA 2 and mtCA 3, respectively. Inhibitory activity of acetazolamide and methazolamide against mtCA 1 and mtCA 3, with inhibition constants in the submicromolar ranges has been reported.26) This promising result may lead to the refurbishment of old carbonic anhydrase inhibitors as new antimycobacterials.
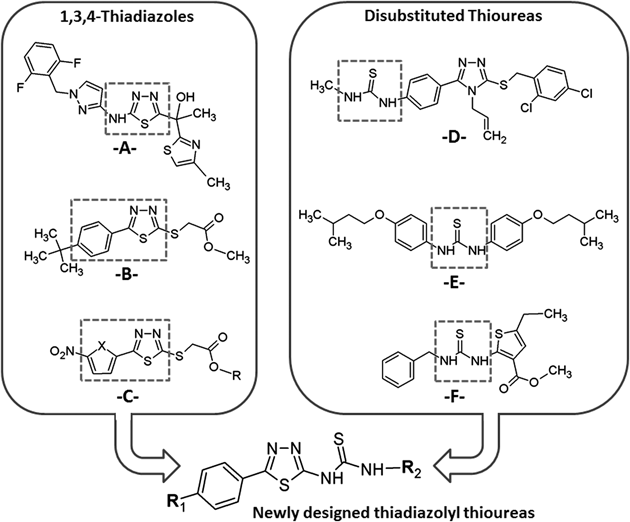
In 2011, GlaxoSmithKline has reported a 1,3,4-thiadiazole derivative containing pyrazole and thiazole linkages. This scaffold [A] has been reported to show potent activity towards Mtb (minimum inhibitory concentration (MIC)=0.19 µM) via potent enoyl acyl carrier protein reductase (InhA) inhibition (IC50=3 nM)27,28) (Fig. 1). Two other reports reveal antituberculosis properties of 1,3,4-thiadiazole scaffolds [B and C] with aryl substitution at C5 position.29,30) On the other hand, antimycobacterial potency of thioureas such as isoxyl [E] is well documented. A thiophenyl–thiourea derivative [F] has been found as a potent inhibitor of mycobacterial growth (IC90=0.23 µg/mL).29) In a previous report, we identified a thiourea derivative linked with 3-alkylthio-1,2,4-triazole moiety [D] as an inhibitor of M. tuberculosis H37Rv though it has a low selectivity.7)
Considering the findings above and in continuation of our efforts for the development of anti-infective agents, we undertook the design and synthesis of some novel prototypes which possess advantage of the two pharmacophores of thiourea and 1,3,4-thiadiazole in single molecular backbone. Our design strategy included to combine the antimycobacterial pharmacophores (indicated in dashed boxes), a 2-amino/5-aryl substituted 1,3,4-thiadiazole ring [A, B, C] with aryl/heteroaryl thiourea moiety [D, E, F], as illustrated in Fig. 2. During the course of this study, we synthesized and characterized hybrid compounds comprising thiourea and 1,3,4-thiadiazole motifs and evaluated them for their anti-tuberculosis activity against M. tuberculosis H37Rv strain, whereas their cytotoxicity profile was assayed by using Vero cells.
MATERIALS AND METHODSSynthetic ChemistryAll solvents and reagents were obtained from commercial sources and used without purification. All melting points (°C, uncorrected) were determined using Kleinfeld SMP-II basic model melting point apparatus. Elemental analyses were obtained using Elementar Analysensysteme GmbH varioMICRO CHNS and are consistent with the assigned structures. Infrared spectra were recorded on a Shimadzu FTIR 8400S and data are expressed in wavenumber ν (cm−1). NMR spectra were recorded on Bruker AVANCE-DPX 400 at 400 MHz for 1H-NMR and 100 MHz for 13C-NMR (decoupled), the chemical shifts were expressed in δ (ppm) downfield from tetramethylsilane (TMS) using dimethyl sulfoxide (DMSO)-d6 as solvent. High resolution (HR) electron impact (EI) and FAB-MS was recorded on a Jeol JMS-700 instrument. HR electrospray ionization (ESI)-MS was recorded on a ICR Apex-Qe instruments. The liquid chromatographic system consists of an Agilent technologies 1100 series instrument equipped with a quaternary solvent delivery system and a model Agilent series G1315 A photodiode array detector. A Rheodyne syringe loading sample injector with a 50 µL sample loop was used for the injection of the analytes. Chromatographic data were collected and processed using Agilent Chemstataion Plus software. The separation was performed at ambient temperature by using a reversed phase Kromasil 5C-18 (4.6×250 mm, 5 µm particle size) column. All experiments were performed in gradient mode. The mobile phase was prepared by mixing acetonitrile and bidistilled water (50 : 50 v/v during 0–3 min, 75 : 25 v/v during 3–5 min, 100 : 0 v/v during 5–7 min, 100 : 0 v/v during 7–12 min, 75 : 25 v/v during 12–15 min, 50 : 50 v/v during 15–18 min) and filtered through a 0.45 µm pore filter and subsequently degassed by ultrasonication, prior to use. Solvent delivery was employed at a flow rate of 1 mL·min−1. Detection of the analytes was carried out at 254 and 280 nm.
Synthesis of 1-Aroylthiosemicarbazides 1, 2 were carried out according to the procedure given in lit24); 4-chlorobenzoylthiosemicarbazide 1 (melting point (mp): 214°C lit31): 218–220°C), 4-fluorobenzoylthiosemicarbazide 2 (mp: 180°C lit31): 172°C).
Synthesis of 2-Amino-5-(4-substituted phenyl)-1,3,4-thiadiazoles 3, 4 were performed according to the literature method24); 2-amino-5-(4-chlorophenyl)-1,3,4-thiadiazole 3 (mp: 230°C lit32): 226–227°C), 2-amino-5-(4-fluorophenyl)-1,3,4-thiadiazole 4 (mp: 240°C lit33): 232–234°C).
Synthesis of 1-[5-(4-Chloro/4-fluorophenyl)-1,3,4-thiadiazole-2-yl]-3-substituted Thioureas 5–20The solution of 5-(4-chloro/4-fluorophenyl)-1,3,4-thiadiazol-2-amine (3, 4) in acetonitrile was reacted with equimolar amounts of appropriate isothiocyanates at 140°C for 20 h. Acetonitrile was evaporated under vacuo and the solid precipitated was washed with HCl 5% at 70°C and then recrystallized from appropriated solvents.
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-[2-(morpholine-4-yl)ethyl]thiourea (5)1H-NMR (400 MHz, DMSO-d6) δ: 2.43–2.55 (4H, m), 3.47–3.85 (8H, m), 7.57 (2H, d, J=8.5 Hz), 7.92 (2H, d, J=8.5 Hz), 8.47 and 8.61 (1H, br s and br s), 12.37 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 53.35 and 53.72 (d), 56.47 (s), 65.45 (s), 66.85 (s), 128.79 (s), 129.86 (s), 130.07 (s), 135.64 (s), 157.66 (s), 163.96 (s), 180.49 (s). IR, cm−1: 3321, 1654, 1114. HR-MS (EI+) m/z: 383.0649 (Calcd for C15H1835ClN5O32S2: 383.0641), 349.0879 (Calcd for C15H17N5O32S34S: 349.0874). HPLC tR (min): 10.45. mp: 247–249°C (acetonitrile).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-[2-(phenyl)ethyl]thiourea (6)1H-NMR (400 MHz, DMSO-d6) δ: 2.91 (2H, t, J=7.2 Hz), 3.73–3.78 (2H, m), 7.21–7.35 (5H, m), 7.57 (2H, d, J=8.5 Hz), 7.91 (2H, d, J=8.5 Hz), 8.61 (1H, br s), 12.41 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 34.63 (s), 46.43 (s), 126.97 (s), 128.58 (s), 128.98 and 129.04 (d), 129.14 and 129.37 (d), 129.47 and 129.66 (d), 130.05 (s), 130.97 (s), 135.01 and 135.55 (d), 139.65 and 139.98 (d), 180.49 (s). IR, cm−1: 3321, 1641, 1112. HR-MS (EI+) m/z: 374.0473 (Calcd for C17H1535ClN432S2: 374.0426), 340.0526 (Calcd for C17H1335ClN432S : 340.0549). HPLC tR (min): 10.59. mp: 244°C (acetonitrile).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-[2-(4-chlorophenyl)ethyl]thiourea (7)1H-NMR (400 MHz, DMSO-d6) δ: 2.84–2.96 (2H, m), 3.75–3.82 (2H, m), 7.21–7.35 (4H, m), 7.57–7.62 (2H, m), 7.78–7.93 (2H, m), 8.70 (1H, br s), 11.99 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 33.82 (s), 46.09 (s), 128.81 and 128.86 (d), 129.81 (s), 131.09 (s), 131.71 (s), 135.67 (s), 138.55 (s), 180.49 (s). IR, cm−1: 3321, 1641, 1112, 1089. HR-MS (EI+) m/z: 408.0039 (Calcd for C17H1437Cl2N4S2: 408.0037), 374.0152 (Calcd for C17H1237ClN4S: 374.0159). HPLC tR (min): 10.58. mp: 222–223°C (acetonitrile).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(benzoyl)thiourea (8)1H-NMR (400 MHz, DMSO-d6) δ: 7.56–7.64 (4H, m), 7.67–7.72 (1H, m), 8.01–8.04 (3H, m), 8.14 (1H, d, J=7.2 Hz), 12.29 (1H, br s), 13.23 and 14.38 (1H, br s and br s). 13C-NMR (100 MHz, DMSO-d6) δ: 116.99 and 117.28 (d), 127.47 (s), 129.11 and 129.89 (d), 130.01 (s), 132.06 (s), 133.76 (s), 134.21 (s), 160.03 (s), 161.69 (s), 162.38 (s), 181.33 (s). IR, cm−1: 3321, 1670, 1641, 1176. HR-MS (EI+) m/z: 376.0016 (Calcd for C16H1137ClN4OS2: 376.0063), 374.0057 (Calcd for C16H1135ClN4OS2: 374.0063), 105.0315 (Calcd for C7H5O: 105.0334), 77.0422 (Calcd for C6H5: 77.0385). HPLC tR (min): 10.64. mp: 313–315°C (acetonitrile).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-cyanophenyl)thiourea (10)1H-NMR (400 MHz, DMSO-d6) δ: 7.58 (2H, d, J=6.6 Hz), 7.37 (2H, d, J=8.7 Hz), 7.89 (2H, d, J=10.8 Hz), 7.99 (2H, d, J=8.7 Hz), 8.70 (1H, br s), 10.90 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 105.42 (s), 119.93 (s), 121.94 (s), 128.96 (s), 129.44 (s), 130.16 (s), 133.52 (s), 136.29 (s), 144.51 (s), 154.15 (s), 169.52 (s), 185.52 (s). IR, cm−1: 3311, 2205, 1641, 1176. HR-MS (EI+) m/z: 371.0027 (Calcd for C16H1035ClN5S2: 371.0066), 337.0181 (Calcd for C16H835ClN5S: 337,0188), 252.9544 (Calcd for C9H435ClN3S2: 252.9535), 118.0539 (Calcd for C7H6N2: 118.0531). HPLC tR (min): 9.68. mp: 263–264°C (methanol).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-fluorophenyl)thiourea (11)1H-NMR (400 MHz, DMSO-d6) δ: 7.16 (2H, t, J=8.4 Hz), 7.49–7.76 (4H, m), 7.88–7.97 (3H, m,), 10.56 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 115.57 and 115.87 (d), 128.59 (s), 128.89 (s), 129.64 (s), 130.13 (s), 136.06 (s), 136.62 (s), 180.49 (s). IR, cm−1: 3327, 1653, 1637, 1229, 1176. HR-MS (EI+) m/z: 364.0007 (Calcd for C15H1035ClFN4S2: 364.0019), 330.0129 (Calcd for C15H835ClFN4S: 330.0142), 252.9545 (Calcd for C9H435ClN3S2: 252.9535), 111.0471 (Calcd for C6H6NF: 111.0484). HPLC tR (min): 9.95. mp: 283–284°C (methanol).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(3,5-bistrifluoromethylphenyl)thiourea (12)1H-NMR (400 MHz, DMSO-d6) δ: 7.47–7.52 (2H, m), 7.57–7.61 (1H, m), 7.72–7.76 (2H, m), 7.84 (1H, s), 7.89–7.95 (1H, m), 8.17 and 8.44 (1H, s and s), 10.99 and 11.65 (1H, s and s). 13C-NMR (100 MHz, DMSO-d6) δ: 118.31 (s), 121.94 (s), 122.36 (s), 125.75 (s), 128.58 (s), 129.86 (s), 130.86 (s), 131.50 (s), 134.67 (s), 136.32 (s), 141.76 (s), 155.84 (s), 169.53 (s), 189.45 (s). IR, cm−1: 3213, 1630, 1176. HR-MS (EI+) m/z: 481.9849 (Calcd for C17H935ClF6N4S2: 481.9861), 447.9963 (Calcd for C17H735ClF6N4S: 447.9984). HPLC tR (min): 11.54. mp: 296–299°C (methanol).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(5-chloro-2-methylphenyl)thiourea (13)1H-NMR (400 MHz, DMSO-d6) δ: 2.15 (3H, s), 7.21–7.36 (3H, m), 7.55–7.76 (3H, m), 7.86 (2H, d, J=8.4 Hz), 10.14 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 18.00 (s), 128.92 (s), 129.64 (s), 130.11 (s), 130.46 (s), 134.01 (s), 132.52 (s), 135.99 (s), 139.74 (s), 189.45 (s). IR, cm−1: 3313, 1641, 1176. HR-MS (EI+) m/z: 393.9869 (Calcd for C16H1235Cl2N4S2: 393.9880), 360.0010 (Calcd for C16H1035Cl2N4S: 360.0003). HPLC tR (min): 10.64. mp: 284–286°C (methanol).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(2-chloro-5-trifluoromethylphenyl)thiourea (14)1H-NMR (400 MHz, DMSO-d6) δ: 7.56–7.65 (4H, m), 7.76 (1H, d, J=8.7 Hz), 7.87–7.98 (3H, m), 10.33 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 122.42 (s), 124.11 (s), 125.10 (s), 126.03 (s), 126.84 (s), 128.44 (s), 129.53 (s), 130.15 (s), 135.34 (s), 136.12 (s), 138.02 (s), 180.49 (s). IR, cm−1: 3286, 3204, 1634, 1121. HR-MS (FAB) m/z: 448.9626 (Calcd for C16H1035Cl2F3N4S2: 448.9670), 414.9787 (Calcd for C16H835Cl2F3N4S: 414.9793). HPLC tR (min): 11.30. mp: 306–309°C (methanol).
1-[5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-chloro-3-trifluoromethylphenyl)thiourea (15)1H-NMR (400 MHz, DMSO-d6) δ: 7.53–7.70 (3H, m), 7.85 (2H, d, J=8.7 Hz), 8.08–8.16 (2H, m), 9.61 (1H, br s), 10.84 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 117.98 (s), 121.26 and 121.59 (d), 124.3 and 124.92 (d), 125.22 (s), 128.55 and 128.84 (d), 129.20 and 129.44 (d), 129.66 and 129.82 (d), 130.03 (s), 131.01 (s), 132.23 and 132.69 (d), 136.21 (s), 139.77 (s), 154.09 (s), 185.32 (s). IR, cm−1: 3327, 1653, 1136, 1121. HR-MS (FAB) m/z: 448.9673 (Calcd for C16H1035Cl2F3N4S2: 448.9670), 414.9826 (Calcd for C16H835Cl2F3N4S: 414.9793). HPLC tR (min): 11.40. mp: 298–302°C (methanol).
1-[5-(4-Fluorophenyl)-1,3,4-thiadiazole-2-yl]-3-benzoylthiourea (16)1H-NMR (400 MHz, DMSO-d6) δ: 7.36–7.64 (5H, m), 8.01–8.11 (4H, m), 12.31 (1H, br s), 13.15 (1H, br s). 13C-NMR (100 MHz, DMSO-d6) δ: 116.99 and 117.28 (d), 127.47 (s), 129.11 and 129.89 (d), 130.01 (s), 132.06 (s), 133.76 (s), 134.21 (s), 160.03 (s), 161.69 and 162.38 (d), 165.68 (s), 181.33 (s). IR, cm−1: 3306, 1672, 1635, 1220, 1153. HR-MS (ESI) m/z: 359.0433 (Calcd for C16H12FN4OS2: 359.0431). HPLC tR (min): 8.75. mp: 262°C (methanol).
1-[5-(4-Fluorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-cyanophenyl)thiourea (18)1H-NMR (400 MHz, DMSO-d6) δ: 7.37 (2H, t, J=8.1 Hz), 7.73 (2H, d, J=8.7 Hz), 7.93–8.02 (5H, m), 10.88 (1H, s). 13C-NMR (100 MHz, DMSO-d6) δ: 105.37 (s), 117.08 and 117.38 (d), 121.92 (s), 127.16 and 127.20 (d), 129.63 and 129.75 (d), 133.52 (s), 144.54 (s), 162.58 (s), 165.89 (s), 180.49 (s). IR, cm−1: 3313, 2207, 1658, 1234, 1157. HR-MS (ESI) m/z: 356.0437 (Calcd for C16H11FN5S2: 356.0434). HPLC tR (min): 8.53. mp: 281–283°C (methanol).
1-[5-(4-Fluorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-fluorophenyl)thiourea (19)1H-NMR (400 MHz, DMSO-d6) δ: 7.16 (2H, t, J=7.8 Hz), 7.36 (3H, t, J=8.7 Hz), 7.64–7.68 (2H, m), 7.91–8.01 (2H, m), 10.55 (1H, s). 13C-NMR (100 MHz, DMSO-d6) δ: 115.57 and 115.86 (d), 117.03 and 117.33 (d), 127.36 (s), 129.53 and 129.65 (d), 136.63 (s), 162.46 (s), 165.76 (s), 180.49 (s). IR, cm−1: 3325, 3201, 1651, 1224, 1155. HR-MS (ESI) m/z: 349.0392 (Calcd for C15H11F2N4S2: 349.0387). HPLC tR (min): 9.02. mp: 293–295°C (methanol).
1-[5-(4-Fluorophenyl)-1,3,4-thiadiazole-2-yl]-3-(4-chloro-3-trifluoromethylphenyl)thiourea (20)1H-NMR (400 MHz, DMSO-d6) δ: 7.37–7.63 (4H, m), 7.92–7.97 (3H, m), 8.13 (1H, s), 10.84 (1H, s). 13C-NMR (100 MHz, DMSO-d6) δ: 31.39 (s), 117.08 and 117.37 (d), 127.24 (s), 129.61 and 129.73 (d), 132.29 (s), 139.80 (s), 162.56 (s), 207.24 (s). IR, cm−1: 3329, 3225, 1651, 1226, 1159. HR-MS (ESI) m/z: 432.9972 (Calcd for C16H11ClF4N4S2: 432.9966). HPLC tR (min): 10.21. mp: 299–301°C (methanol).
Anti-tuberculosis ActivityThe antituberculosis activity of compounds was tested against M. tuberculosis H37Rv strain. For the MIC determination, the compounds were dissolved in DMSO and serial two fold dilutions were done in Middlebrook 7H9 broth with glycerol. Microorganisms were suspended in Middlebrook 7H9 broth to match the turbidity of 0.5 McFarland (1.5×108 cfu/mL) and 1/10 dilution was prepared from this suspension and used as inoculum. The tested final concentrations ranged between 512 to 0.5 µg/mL. To make sure that DMSO did not show any inhibitory activity, controls prepared with serial dilutions of DMSO were also tested. Tubes were incubated at 37°C for 24 h and then examined for turbidity. MIC was determined if turbidity was observed in the positive control tube containing no compound and no turbidity in the negative control tube containing no microorganism.34–36)
Antimicrobial ActivityThe antimicrobial activities of all compounds were evaluated in the Department of Genetics and Bioengineering, Faculty of Engineering and Architecture, Yeditepe University. The antibacterial and antifungal activities of the compounds were evaluated against 8 microbial cultures isolates of 6 bacteria and 1 yeast species by micro-well dilution assay as described below.37) Microorganisms were provided by the Department of Genetics and Bioengineering, Faculty of Engineering and Architecture, Yeditepe University, Istanbul, Turkey. The microorganisms used were Staphylococcus aureus, Escherichia coli, Bacillus subtilis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus epidermidis and Candida albicans. Ampicillin, Cefepime and Amphotericin B were used as the positive sensitivity reference standard for bacteria and yeast.
Micro-well Dilution AssayThe sensitivity of the bacterial strains towards the compounds was quantitatively evaluated from the MIC values obtained by the micro-well dilution method. The inocula of the bacterial strains were prepared from 12-h broth cultures and suspensions were adjusted to 0.5 McFarland standard turbidity. Compounds dissolved in DMSO were first prepared at the highest concentration to be tested (200 µg/mL), and then serial two-fold dilutions were made in order to obtain a concentration range from 6.25 to 200 µg/mL, in 15-mL sterile test tubes containing nutrient broth. The 96-well plates were prepared by dispensing into each well 95 µL of nutrient broth and 5 µL of the inoculum. Two hundred microliters of nutrient broth without inoculum was transferred into the first wells as positive control. Aliquots, (100 µL) taken from the 200-µg/mL stock solution, were added to the second well.
One hundred microliters from the respective serial dilutions was transferred into 5 consecutive wells. The last well containing 195 µL of nutrient broth without compound and 5 µL of the inoculum on each strip was used as negative control. Contents of each well were mixed on plate shaker at 300 rpm for 20 s and then incubated at appropriate temperatures for 24 h. Microbial growth in each medium was determined by reading the absorbance (Abs) at 630 nm using the ELx 800 universal microplate reader (Biotek Instrument Inc., Highland Park, VT, U.S.A.) and confirmed by plating 5-µL samples from clear wells on nutrient agar medium. The MIC was defined as the lowest concentration of the compounds to inhibit the growth of microorganisms. Cefepime (Maxipime®, Bristol-Myers Squibb, Istanbul, Turkey) at the concentration range of 500–7.8 µg/mL was prepared in nutrient broth and used as standard drug for positive control.
In Vitro Antiviral AssaysA detailed account of experimental data related to in vitro antiviral assays (inhibition of human immunodeficiency virus (HIV)-induced cytopathicity in MT-4 cells, antiviral assays other than HIV and anti-influenza assays) were given in literature.38)
RESULTS AND DISCUSSION
Chemistry4-Chlorobenzoylthiosemicarbazide 1, 4-fluorobenzoylthiosemicarbazide 2, their corresponding 1,3,4-thiadiazoles; 2-amino-5-(4-chlorophenyl)-1,3,4-thiadiazole 3 and 2-amino-5-(4-fluorophenyl)-1,3,4-thiadiazole 4, were synthesized according to the procedures given in literature.24) Their purity were checked by TLC and HPLC and melting points of compounds 1–4 are found to be in accordance with literature.31–33) Synthetic route to compounds 5–20 is presented in Chart 1. As the final step, 5-(4-substituted phenyl)-1,3,4-thiadiazol-2-amines (3, 4) were reacted with appropriate isothiocyanates in acetonitrile to yield 1-[5-(4-substituted phenyl)-1,3,4-thiadiazole-2-yl]-3-substituted thioureas 5–20. The purity of the synthesized compounds were checked by TLC and HPLC. Since synthesis procedure and structural characterization data of compounds 9 and 17 were already reported by George et al.,39) spectral data were not provided for these compounds.
HR-MS confirmed the molecular weights and empirical formulae of compounds 5–20, with less than 5 mmu bias between calculated and experimental m/z values of molecular ions. Ionization mode was EI for compounds 5–13. Compounds 14 and 15 which were analysed using FAB procedure and they demonstrated MH+ peaks instead of molecular ion (M+) peaks. Compounds 16–20 were also analysed using another soft ionization technique; ESI procedure and their MH+ peaks were determined. The mass of M+ peaks and experimental fragments of compounds 5–13 and MH+ peaks of compounds 14–20 match the mass of corresponding calculated ones. Compounds 5–13 cleave off H2S forming carbodiimide fragments in accordance with literature.40) Compounds 10 and 11 allow unambiguous fragmentations of characteristics product ions for thiourea based compounds yielding the common 2-(4-chlorophenyl)-5-isothiocyanato-1,3,4-thiadiazole fragment at m/z 252.9544 and as well as 4-aminobenzonitrile fragment at m/z 118.0539 and 4-fluoroaniline fragment at m/z 111.0471, respectively.8,41)
The diagnostic N–H strecthing vibrations of thiourea compunds 5–20 appeared in 3128–3361 cm−1 region.42) This wide range and varying number of N–H bands may be attributable to intramolecular hydrogen bonds.43) Since a S–H vibration at approximately 2600 cm−1 has not been detected, we may indicate that our thiourea compounds remain in thioketo-amine form in solid state. Fourier transform-infrared (FT-IR) spectra revealed C=S strecthing vibrations at 1112–1176 cm−1 region.38)
The characteristic N2–H signals of thiourea compounds noted as a singlet or two singlets corresponding to one proton at the region of 10.14–12.41 ppm. An other characteristic signal N1–H was determined either at aromatic region or between 8.13–9.61 ppm due to their intra and/or intermolecular hydrogen bond formation tendency by means of equilibria between syn and anti conformations.44,45) In the 1H-NMR spectra of compounds 8 and 16 with benzoyl moiety on thiourea structure, N1–H was determined at 12.29 and 12.31 ppm, respectively and N2-H was determined at 13.23 and 14.38 and 13.15 ppm, respectively. 13C-NMR data of representative derivatives was found to be descriptive for carbon framework and detection of C=S signal between 180–207 ppm was evaluated as an evidence for thiourea formation. Meanwhile the aromatic carbon resonances due to phenyl and 1,3,4-thiadiazole rings were also observed in expected regions.24,25,39)
Antituberculosis ActivityAll synthesized compounds 5–20 were initially screened for their in vitro antituberculosis activity against M. tuberculosis H37Rv strain. The MIC vs. M. tuberculosis H37Rv was determined by a broth microdilution method as previously described (Table 1). Minimum cytotoxic concentration (MCC) on Vero cells was also determined to evaluate the selectivity of the synthesized compounds. Antituberculosis activity and cytotoxicity results of compounds 5–20 were compared to isoniazid (INH) and ethambutol which were used as reference drugs (Table 1).
Table 1.
In Vitro Antimycobacterial Activity of Compounds
5–
20 against
M. tuberculosis H
37Rv
 |
|---|
| Laboratory code | Compd ID | R1 | R2 | MIC (µM) | MCCa) (µM) | SI (MCC/MIC) | PASS predictionc) |
|---|
| Pa | Pi |
|---|
| KUC060101 | 5 | Cl | 2-(Morpholin-1-yl)ethyl | 83.78 | >100 | >1.2 | 0.364 | 0.043 |
| KUC060102 | 6 | Cl | 2-Phenylethyl | 170.70 | 100 | 0.6 | 0.469 | 0.016 |
| KUC060103 | 7 | Cl | 2-(4-Chlorophenyl)ethyl | 78.17 | 100 | 1.3 | 0.468 | 0.016 |
| KUC060104 | 8 | Cl | Benzoyl | 170.72 | >100 | >0.6 | 0.658 | 0.005 |
| KUC060105 | 9 | Cl | Phenyl | 46.13 | ≥20 | ≥0.4 | 0.684 | 0.004 |
| KUC060106 | 10 | Cl | 4-Cyanophenyl | 43.02 | ≥20 | ≥0.5 | 0.521 | 0.010 |
| KUC060107 | 11 | Cl | 4-Fluorophenyl | 10.96 | ≥20 | ≥1.8 | 0.582 | 0.006 |
| KUC060108 | 12 | Cl | 3,5-Bis(trifluoromethyl)phenyl | 33.14 | 20 | 0.6 | 0.563 | 0.007 |
| KUC060109 | 13 | Cl | 5-Chloro-2-methylphenyl | 80.94 | 20 | 0.2 | 0.638 | 0.005 |
| KUC060110 | 14 | Cl | 2-Chloro-5-(trifluoromethyl)phenyl | 71.22 | 20 | 0.3 | 0.483 | 0.014 |
| KUC060111 | 15 | Cl | 4-Chloro-3-(trifluoromethyl)phenyl | 17.81 | 100 | 5.6 | 0.538 | 0.009 |
| KUC060113 | 16 | F | Benzoyl | 178.56 | 100 | 0.6 | 0.578 | 0.007 |
| KUC060114 | 17 | F | Phenyl | 48.42 | 100 | 2.1 | 0.611 | 0.005 |
| KUC060115 | 18 | F | 4-Cyanophenyl | 45.02 | 100 | 2.2 | 0.450 | 0.019 |
| KUC060116 | 19 | F | 4-Fluorophenyl | 11.48 | 100 | 8.7 | 0.610 | 0.005 |
| KUC060118 | 20 | F | 4-Chloro-3-(trifluoromethyl)phenyl | 36.96 | 4 | 0.1 | 0.458 | 0.018 |
| Isoniazid | | | | 0.073–1.45 | NDb) | NA | 0.813 | 0.003 |
| Ethambutol | | | | 6.12–24.47 | NDb) | NA | 0.926 | 0.002 |
a) Minimum cytotoxic concentration was determined on Vero cells. b) Both isoniazid and ethambutol were reported to be non-toxic at 62.5 µg/mL (equivalent to 455.74 µM for isoniazid and 305.91 µM for ethambutol) on Vero cells.46) c) Antituberculosis activity prediction (Pa: probability of activity ; Pi: probability of inactivity).
Compounds having either 4-chlorophenyl (compounds 5–15) or 4-fluorophenyl (compounds 16–20) substituents on 1,3,4-thiadiazole ring were found to be active in varying levels. Three compounds (11, 15, and 19) from this series can be distinguished from others by their promising activity profiles by their MIC values comparable to ethambutol. Compounds 11 and 19 were appreciated as the most active representatives with the MIC values of 10.96 and 11.48 µM, respectively, whereas compound 19 was confirmed to possess higher selectivity than compound 11. Compound 15 was shown to inhibit M. tuberculosis H37Rv strain with the MIC values of 17.81 µM. Except for 11, 15 and 19, antituberculosis activity of the other compounds were observed between 33.14–178.56 µM. The compounds 9, 10, 12, 17, 18, 20 with MIC values between 33.14 and 48.42 µM are assumed to possess mild antituberculosis activity; and the compounds 6–8, 13, 14, 16 with MIC values greater than 70 µM are considered as weakly active against M. tuberculosis H37Rv strain.
The nature of the substituents on either 1,3,4-thiadiazole (R1) and thiourea (R2) was found to affect the activity of the compounds 5–20. Introduction of 4-fluorophenyl substitution on N3 of thiourea moiety yielded the most selective activity (compound 19). Replacing 4-fluorophenyl substitution on N3 of thiourea moiety by 4-chloro-3-trifluoromethylphenyl moiety (compound 20) reduced the activity by 3.2-fold as compared to lead 19. 5-(4-Chlorophenyl)-1,3,4-thiadiazole and 5-(4-fluorophenyl)-1,3,4-thiadiazole were introduced to N1 of thiourea moiety in order to obtain the optimal substitution pattern, consequently 5-(4-fluorophenyl)-1,3,4-thiadiazole moiety seemed to contribute more to the antituberculosis activity than the others.
Active compounds 15 and 19 were observed to be non-toxic at 100 µM, whereas compound 11 showed toxicity at ≥20 µM. Standard drugs, isoniazid and ethambutol were reported to be non-toxic up to 62.5 µg/mL (equivalent to 455.7 µM for isoniazid and 305.91 µM for ethambutol) on Vero cells.46) In another work, IC50 values of isoniazid and ethambutol were reported to be higher than 455.7 and 225.4 µM, respectively.47) Thus, isoniazid and ethambutol appeared as safer antituberculosis agents compared to compounds 11, 15 and 19.
Antibacterial ActivityThe antibacterial activity of compounds 5–20 except for 6 and 11 was tested against Staphylococcus aureus, Escherichia coli, Bacillus subtilis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus epidermidis and Candida albicans. The MIC was determined by a micro well dilution method (Table 2). Compounds 12 and 14 were found as the most active compounds with the MIC values of 0.19 and 0.23 µg/µL against B. subtilis. The other compounds were not found active against specifically and significantly active against bacteria mentioned above.
Table 2.
In Vitro Antibacterial and Antifungal Activity of Compounds
5–
20 (MIC in µg/µL)
| Compd ID | E. coli | S. aureus | B. subtilis | K. pneumoniae | P. aeruginosa | S. epidermidis | C. albicans |
|---|
| 5 | 1.29 | 0.64 | 1.29 | 1.29 | 1.29 | 1.29 | 1.29 |
| 7 | 1.74 | 1.74 | 1.74 | >3.48 | 1.74 | 1.74 | 1.74 |
| 8 | >3.05 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 | 1.53 |
| 9 | 1.35 | 2.70 | 1.35 | 1.35 | 1.35 | 1.35 | 0.68 |
| 10 | 3.73 | 3.73 | 3.73 | 3.73 | 3.73 | 3.73 | 3.73 |
| 12 | 1.49 | 0.74 | 0.19 | 1.49 | 1.49 | 0.74 | 1.49 |
| 13 | 1.68 | 1.68 | 1.68 | 1.68 | 3.35 | 1.68 | 3.35 |
| 14 | 1.80 | 1.80 | 0.23 | 3.60 | 0.90 | 0.90 | 1.80 |
| 15 | 1.73 | 3.73 | 0.86 | 3.45 | 1.73 | 3.73 | 1.73 |
| 16 | 1.86 | 3.73 | 3.73 | 3.73 | 1.86 | 0.93 | 3.73 |
| 17 | 1.78 | >3.55 | >3.55 | 3.55 | 1.78 | >3.55 | 1.78 |
| 18 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 |
| 19 | >3.48 | >3.48 | >3.48 | >3.48 | >3.48 | >3.48 | >3.48 |
| 20 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 | >2.90 |
Depending on antituberculosis and antibacterial activity evaluation results we may propose that our compounds are selectively active against M. tuberculosis H37Rv strain.
Antiviral Activity and Cytotoxicity StudiesThiourea derivatives 5–20 were also evaluated for their broad spectrum antiviral activity against a wide range of DNA and RNA viruses. For each compound, the MIC and the minimal cytotoxic concentration (MCC) or the 50% cytotoxic concentration (CC50) were obtained. Anti-HIV activity and cytotoxicity data were obtained with the synthesized compounds 5–20 using the strains HIV-1 (IIIB) and HIV-2 (ROD) in an MT-4/3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) based assay. None of the synthesized compounds showed any specific activity against HIV-1 (IIIB) or HIV-2 (strain ROD) in MT-4 cells. The compounds were also evaluated for in vitro antiviral activity against herpes simplex virus [HSV-1 (strain KOS), thymidine kinase deficient (TK−) strain of HSV-1 resistant to acyclovir (ACVr), HSV-2 (G)], vaccinia virus (VV) and vesicular stomatitis virus (VSV) in HEL cell cultures ; VSV, Coxsackie virus B4 and respiratory syncytial virus (RSV) in HeLa cell cultures ; Parainfluenza-3 virus, Reovirus-1, Sindbis virus, Coxsackie virus B4 and Punta Toro virus in Vero cell cultures; influenza A (H1N1, H3N2) influenza B virus in Madin–Darby canine kidney (MDCK) cell cultures; feline coronavirus (FIPV) and feline herpes virus in CRFK cell cultures.
As a result of antiviral screening of 5–20, none of the evaluated compounds showed specific antiviral effects (i.e., minimal antivirally effective concentration >5-fold lower than minimal cytotoxic concentration) against any of the viruses examined (data not shown).
Prediction of Drug-Likeness and Absorption, Distribution, Metabolism, and Excretion (ADME) Properties of 5–20In the development of new drug candidates intended for oral use, oral bioavailability and proper drug delivery are considered to play an important role.48) About one-third of drug candidates fail during development due to their poor pharmacokinetic profiles.49) As an ADME property, a low and variable bioavailability is the major reason to quit further development of the drug candidate.50) Therefore, a computational study for prediction of ADME properties of the molecules was performed by determination of lipophilicity, topological polar surface area (TPSA), absorption (% ABS) and simple molecular descriptors used by Lipinski in formulating his “rule of five.”51) Calculations were performed using Molinspiration online property calculation toolkit.52) Table 3 represents a calculated percentage of absorption (% ABS), topological polar surface area (TPSA) and Lipinski parameters of the compounds 5–20. Percentage of absorption (% ABS) was estimated using the equation: % ABS=109−(0.345×TPSA), according to Zhao et al.53) TPSA was also calculated using Molinspiration online property calculation toolkit according to the fragment-based method of Ertl et al.54) Polar surface area, together with lipophilicity, is an important property of a molecule in transport across biological membranes. Too high TPSA values give rise to a poor bioavailability and absorption of a drug. According to the above criterions, calculated percentages of absorption for compounds 5–20 ranged between 84 and 92%.
Table 3. Predicted ADME, Lipinski Parameters and Molecular Properties of the Synthesized Compounds
5–
20a)| Compd ID | MW | Volume | TPSA | %ABS | n-ROTB | n-ON | n-OHNH | mi Log P | n Violations |
|---|
| 5 | 381.958 | 323.608 | 53.076 | 91 | 7 | 5 | 2 | 4.237 | 0 |
| 6 | 374.922 | 309.065 | 49.838 | 92 | 7 | 4 | 2 | 5.103 | 1 |
| 7 | 409.367 | 322.601 | 49.838 | 92 | 7 | 4 | 2 | 5.781 | 1 |
| 8 | 374.878 | 294.445 | 66.909 | 86 | 5 | 5 | 2 | 4.333 | 0 |
| 9 | 346.868 | 275.462 | 49.838 | 92 | 5 | 4 | 2 | 4.360 | 0 |
| 10 | 371.878 | 292.321 | 73.630 | 84 | 5 | 5 | 2 | 4.115 | 0 |
| 11 | 364.858 | 280.393 | 49.838 | 92 | 5 | 4 | 2 | 4.523 | 0 |
| 12 | 482.862 | 338.057 | 49.838 | 92 | 7 | 4 | 2 | 6.078 | 1 |
| 13 | 395.34 | 305.559 | 49.838 | 92 | 5 | 4 | 2 | 5.414 | 1 |
| 14 | 449.31 | 320.295 | 49.838 | 92 | 6 | 4 | 2 | 5.861 | 1 |
| 15 | 449.31 | 320.295 | 49.838 | 92 | 6 | 4 | 2 | 5.861 | 1 |
| 16 | 358.423 | 285.841 | 66.909 | 86 | 5 | 5 | 2 | 3.818 | 0 |
| 17 | 330.413 | 266.857 | 49.838 | 92 | 5 | 4 | 2 | 3.845 | 0 |
| 18 | 355.423 | 283.717 | 73.63 | 84 | 5 | 5 | 2 | 3.600 | 0 |
| 19 | 348.403 | 271.788 | 49.838 | 92 | 5 | 4 | 2 | 4.009 | 0 |
| 20 | 432.855 | 311.691 | 49.838 | 92 | 6 | 4 | 2 | 5.347 | 1 |
a) % ABS: Percentage of absorption, TPSA: topological polar surface area, n-ON: number of hydrogen bond acceptors, n-OHNH: number of hydrogen bond donors, n-ROTB: number of rotatable bonds. Calculations were performed using Molinspiration online property calculation toolkit (http://www.molinspiration.com).
Number of hydrogen bond donors was constant for all of the compounds, and number of hydrogen bond acceptors varied 4 to 5. Investigation of Lipinski parameters of the synthesized compounds showed that all heterocyclic thiourea derivatives of 2-amino-1,3,4-thiadiazoles, might be considered as drug-like candidates for novel anti-tuberculosis agents, as they obeyed the rule of five without violating more than one of them.
There were no direct correlations observed between simple molecular properties such as log P and anti-tuberculosis activity. Nevertheless, it was notable that two most active derivatives (11 and 19) among compounds 5–20 with 4-fluorophenyl substitution at R2 position, had good calculated absorption values and zero violations to Lipinski rule of five.
Osiris CalculationsPrediction of Toxicity, Solubility, Drug-Likeness and Drug Score for Compounds 5–20Potential toxicity, solubility, and drug-like properties (solubility, drug-likeness, and drug score) of the thioureas 5–20 were estimated by Osiris Property Explorer.55,56) The predicted risks include mutagenic, tumorigenic, irritant and reproductive toxicity (Table 4). The property predictor detects fragments within a given molecule as an indicator for a potential toxicity risk. Toxicity risk alerts are used as indicators showing that the investigated structure may be harmful concerning the specified toxicity risk. From the findings given in Table 4, it can be claimed that the target compounds 5–20 are expected to be free of mutagenic, tumorigenic, irritating (except 14) effects and reproductive toxicity (except 8 and 16). Water solubility of a drug candidate significantly influences its absorption and distribution characteristics. According to the Osiris database, more than 80% of the traded drugs have predicted solubility values greater than −4. As shown in Table 4, the synthesized thioureas 5–20 exhibited solubility values between –2.95 and −6.48.
Table 4. Estimation of Toxicity, Solubility, Drug-Likeness and Drug Score for Thioureas
5–
20| Compound | Toxicity risksa) | Solubility | Drug-likeness | Drug score |
|---|
| Mutagenicity | Tumorigenicity | Irritation | Reproductive |
|---|
| 5 | − | − | − | − | –2.95 | 5.43 | 0.82 |
| 6 | − | − | − | − | −4.90 | 4.62 | 0.55 |
| 7 | − | − | − | − | −5.63 | 4.48 | 0.42 |
| 8 | − | − | − | ++ | −4.91 | 4.58 | 0.35 |
| 9 | − | − | − | − | −4.97 | 2.26 | 0.55 |
| 10 | − | − | − | − | −5.74 | −3.10 | 0.26 |
| 11 | − | − | − | − | −5.28 | 2.49 | 0.51 |
| 12 | − | − | − | − | −6.52 | −18.50 | 0.14 |
| 13 | − | − | − | − | −6.05 | 3.48 | 0.39 |
| 14 | − | − | ++ | − | −6.48 | −4.23 | 0.10 |
| 15 | − | − | − | − | −6.48 | −4.16 | 0.16 |
| 16 | − | − | − | ++ | −4.49 | 2.90 | 0.39 |
| 17 | − | − | − | − | −4.55 | 0.53 | 0.54 |
| 18 | − | − | − | − | −5.32 | −4.83 | 0.29 |
| 19 | − | − | − | − | −4.86 | 1.93 | 0.57 |
| 20 | − | − | − | − | −6.06 | −4.49 | 0.19 |
| Thiacetazone | ++ | − | − | − | −3.24 | 4.23 | 0.53 |
| Isoxyl | − | − | + | – | −5.76 | 1.23 | 0.26 |
a) −: low risk; +: moderate risk; ++: high risk.
Drug-likeness can be described as a complicated balance of diverse molecular properties and structural characteristics indicating whether a given molecule is similar to the common drugs or not.57) Osiris property explorer was also utilized to calculate the fragment-based drug-likeness of the synthesized compounds. A positive value demonstrates that the designed compound contains predominatly fragments which are commonly available in traded drugs.
Results presented in Table 4 indicates that majority of 1,3,4-thiadiazolyl thioureas 5–20 have positive drug-likeness values. The drug score unites porperties such as drug-likeness, c Log P, solubility, molecular weight, and toxicity risks in one useful parameter that may be used to estimate the compound’s whole potential to qualify for a drug.55) A drug score of 0.5 or higher makes the compound a promising lead for further development to reach safe and efficient drugs. The overall drug score values for the thioureas 5–20 were calculated and compared to those of thiacetazone and isoxyl (Table 4). Compounds 5, 6, 9, 11, 17 and 19 possess good drug score values. Of these, compounds 11 and 19 had also good drug-likeness values and antituberculosis activity.
PASS-Assisted Antituberculosis Activity PredictionThe experimental testing of dozens of millions of organic compounds for thousands biological activities is obviously unachievable, mandating the need for computer methods for the search and optimization of new pharmacologically active compounds.58) Structure-based drug design and ligand-based drug design are the two computer-based approaches of drug design which are currently being used.59) Anti-tuberculosis activity of our thiourea based compounds 5–20 was predicted by using the online version of PASS (Prediction of Activity Spectra for Substances) program.60) PASS online is a software that enables to predict biological activity profile of drug-like organic compounds (with a molecular mass range between 50 and 1250 Da) according to their chemical structures for more than 4000 types of biological activity including pharmacological effects, mechanisms of actions, interactions with biotransformation enzymes, side effects and toxic properties. The prediction is based on analysis of the structure–activity relationships in the training set containing information on the structure and biological activity of more than 300000 organic compounds.61) The chemical structure is represented by the set of descriptors of multilevel neighborhoods of atoms (MNA).62) The average prediction accuracy calculated by the leave-one-out cross-validation procedure for the whole training set and for all represented in it types of biological activity is about 95%.63)
The PASS online program provides ‘two parameters as a list of predicted types of activity: the probability “to be active” (Pa) and the probability “to be inactive” (Pi), which vary from zero to one.’ The possibility of experimentally determining a certain kind of activity increases with rising value of Pa and decreasing value of Pi.61) In analyzing a predicted activity spectrum, if those types of activity are selected, for which Pa>0.9, the expected probability to find inactive compounds in the selected set is very low, but we risk missing about 90% of actually active compounds.61) If only compounds with Pa>0.8 are chosen, the probability to find inactive compounds is still low, but about 80% of active compounds are missed etc. It is also worth emphasizing that the probability Pa primarily projects the resemblance of the structure of a given molecule to the structures of the most characteristic active molecules in the corresponding subset of the training set. Thus, as a rule, there is no direct correlation of the Pa values with quantitative activity characteristics. Essentially, each selection is always a compromise between the preferred novelty of tested molecule and risk to obtain the negative result in biological screening.
Another important aspect of interpreting the prediction results is related to novelty of the analyzed compound. If we limit ourselves only to activity types predicted with the highest values of Pa, the compounds selected by the prediction may prove to be analogs of known pharmacological agents. For example, when Pa>0.7, the chances of finding experimental activity are rather high but the compounds found may be close structural analogs of known drugs. If we select in the range 0.5<Pa<0.7, the chances for detecting experimental activity will be lower but the compounds will be less similar to known pharmaceutical agents. For Pi<Pa<0.5, the chances of detecting experimental activity will be even lower but if the prediction is confirmed, the compound found may prove a parent compound for a new chemical class for the biological activity examined.61)
There are many examples of successful use of PASS approach for finding new pharmacological agents.63–69) Antituberculosis activity were predicted for our compound set 5–20 within the range of 0.364<Pa<0.684. Although no direct correlation were observed between Pa values and MIC values, it was notable that most active three compounds had good Pa scores (Pa>0.5). As expected, INH and ethambutol gave high Pa values in the same prediction model.
Molecular ModelingThe recent reports on most promising 1,3,4-thiadiazole derivatives with highly potent activity towards Mtb have revealed the target mycobacterial enoyl acyl carrier protein reductase (InhA) due to assessment of InhA inhibition values of the mentioned compounds.24,25) We were also inspired by the experimental MIC data that are generally in consistent with the InhA inhibitory activities of 1,3,4-thiadiazole derivatives to perform molecular docking study.24,25) In order to further rationalize the biological results of our compounds, we perform molecular docking studies on selected active (compounds 11, 15, 19) and inactive derivatives (compounds 6 and 17) with InhA of M. tuberculosis.
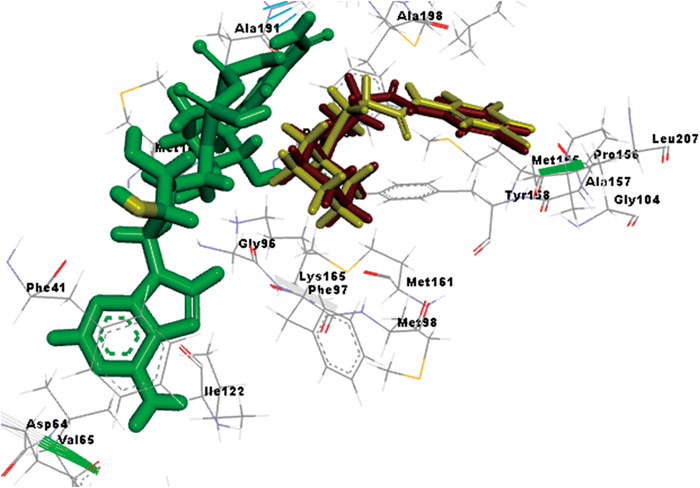
The crystal structure of MTB InhA complexed with reference inhibitor 1-cyclohexyl-N-(3,5-dichlorophenyl)-5-oxopyrrolidine-3-carboxamide (PDB:2H7M) having resolution of 1.62 Å was selected and docking results obtained with Glide, version 5.7, Schrodinger, LLC, New York, NY, 2012. Analysis of the crystal structure of 2H7M revealed that the reference inhibitor in the InhA active site formed hydrogen-bonding network between Tyr158, enzyme active site residues, and the oxidized form of nicotinamide adenine dinucleotide (NAD+) cofactor that probably served as the key feature that governed the orientation of the compound within the active site. Dual hydrogen bonding network was involved with the oxygen atom on the pyrrolidine carbonyl group, InhA catalytic residue Tyr158, and the NAD+. This hydrogen bonding network seemed to be a conserved feature among all the InhA-inhibitor complexes identified so far. The reference 1-cyclohexyl-N-(3,5-dichlorophenyl)-5-oxopyrrolidine-3-carboxamide was re-docked with the active site residues of the MTB InhA to validate the active site cavity. The ligand exhibited highest Glide score of −8.02 kcal/mol and was found in the vicinity of amino acids Tyr158, Phe149, Met199, Ile215, Pro156, Leu218, Met155, Ala211, Ile202, Met103, Leu207, Ala157, Met161, Phe97, Met98, Gly96, Gly104 and Lys165 residues. The re-docking results showed that the compound exhibited similar interactions as that of the original crystal structure with root-mean-square deviation (RMSD) of 0.87 Å suggesting reliability of the docking method. The superimposition of Glide docked conformation of co-crystal with co-crystal of 2H7M is shown in Fig. 3.
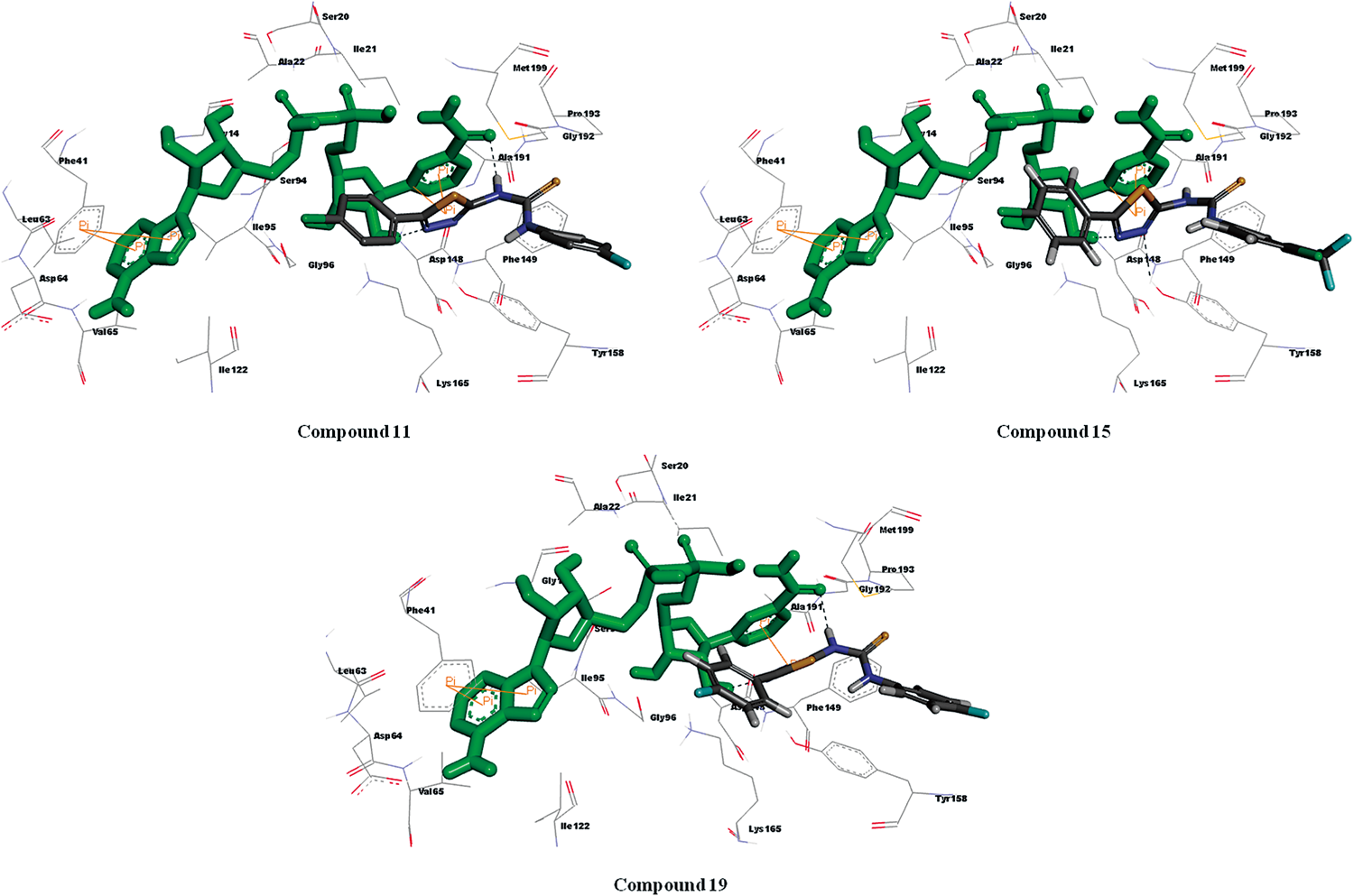
Binding Analysis of Active Compounds (11, 15 and 19)The compounds 11, 15 and 19 with chloro and fluoro group at R1 position and 4-fluorophenyl, 4-chloro-3-(trifluoromethyl)phenyl and 4-fluorophenyl groups at R2 substituent’s, showed good MIC. All the three compounds were docked in the binding site of InhA protein and were displayed good docking scores in the range of −7.12 to −7.83 kcal/mol. The predicted binding pose of the active compounds (11, 15 and 19) suggested that the observed potency may be due to the extensive hydrophobic interactions predicted to be formed with the side chains of Met199, Leu218, Met155, Pro156, Ala157, Ile202, Met103, Phe149 and Met161 along with hydrogen bonding interactions with the ribose hydroxyl group of NAD+ (Fig. 4). Among these three compounds, only compound 15 [R1: Cl; R2: 4-chloro-3-(trifluoromethyl)phenyl] was observed to interact with side chain of Tyr158 via hydrogen bonding between nitrogen atom at 3rd position of 1,3,4-thiadiazole and phenolic group of tyrosine. Apart from these interactions, all the three compounds were further stabilized by Pi–Pi interactions. From the docking results, it was evident that the formation of hydrogen bonds with hydroxyl group of NAD+ along with hydrophobic interactions with the active site were predicted to be the most crucial factors affecting the inhibitory potency of these compounds.
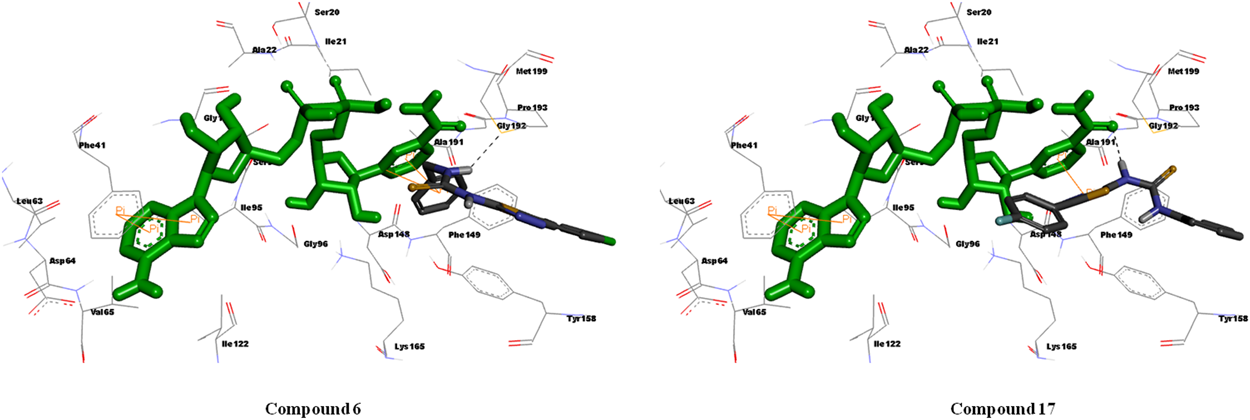
Binding Analysis of Inactive Compounds (6 and 17)Ligand binding analysis of one of the less active derivatives from this subset (6 and 17) showed hydrophobic interactions with some hydrophobic amino acid residues. However, the orientation in the active site cavity of InhA pushed the chloro group in compound 6 and phenyl group in compound 17 away from the cavity, which might be the reason for its lesser activity. In silico analysis of both the compounds indicated that the molecule oriented in a different manner than that of other active derivatives and failed to demonstrate any interaction with Tyr158 residue as well as with NAD+ resulting in less activity in M. tuberculosis strains (Fig. 5). This is well supported by the low docking score of −5.57 and −6.20 kcal/mol.