
39 巻, 4 号
選択された号の論文の27件中1~27を表示しています
- |<
- <
- 1
- >
- >|
Regular Articles
-
2016 年 39 巻 4 号 p. 457-465
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/28 PDF形式でダウンロード (7097K) HTML形式で全画面表示
PDF形式でダウンロード (7097K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 466-472
発行日: 2016/04/01
公開日: 2016/04/01
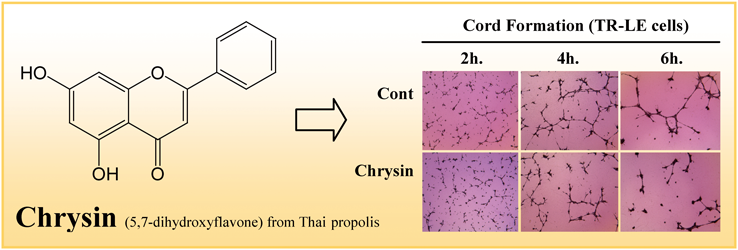 PDF形式でダウンロード (1464K) HTML形式で全画面表示
PDF形式でダウンロード (1464K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 473-483
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (596K) HTML形式で全画面表示
PDF形式でダウンロード (596K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 484-491
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/02/01 PDF形式でダウンロード (908K) HTML形式で全画面表示
PDF形式でダウンロード (908K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 492-501
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (1729K) HTML形式で全画面表示
PDF形式でダウンロード (1729K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 502-515
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (3266K) HTML形式で全画面表示
PDF形式でダウンロード (3266K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 516-523
発行日: 2016/04/01
公開日: 2016/04/01
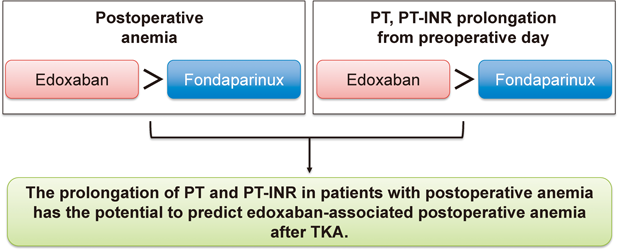 PDF形式でダウンロード (940K) HTML形式で全画面表示
PDF形式でダウンロード (940K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 524-531
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/23 PDF形式でダウンロード (1122K) HTML形式で全画面表示
PDF形式でダウンロード (1122K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 532-539
発行日: 2016/04/01
公開日: 2016/04/01
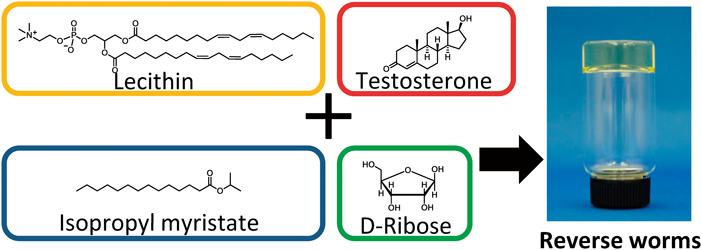 PDF形式でダウンロード (1149K) HTML形式で全画面表示
PDF形式でダウンロード (1149K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 540-546
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (930K) HTML形式で全画面表示
PDF形式でダウンロード (930K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 547-555
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (1477K) HTML形式で全画面表示
PDF形式でダウンロード (1477K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 556-563
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (1503K) HTML形式で全画面表示
PDF形式でダウンロード (1503K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 564-569
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (10235K) HTML形式で全画面表示
PDF形式でダウンロード (10235K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 570-577
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/23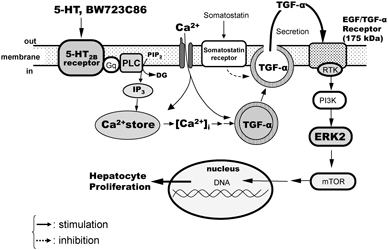 PDF形式でダウンロード (1454K) HTML形式で全画面表示
PDF形式でダウンロード (1454K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 578-586
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (2372K) HTML形式で全画面表示
PDF形式でダウンロード (2372K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 587-592
発行日: 2016/04/01
公開日: 2016/04/01
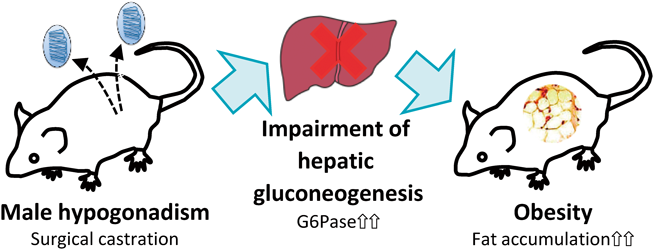 PDF形式でダウンロード (566K) HTML形式で全画面表示
PDF形式でダウンロード (566K) HTML形式で全画面表示 -
Deer Bone Oil Extract Suppresses Lipopolysaccharide-Induced Inflammatory Responses in RAW264.7 Cells2016 年 39 巻 4 号 p. 593-600
発行日: 2016/04/01
公開日: 2016/04/01
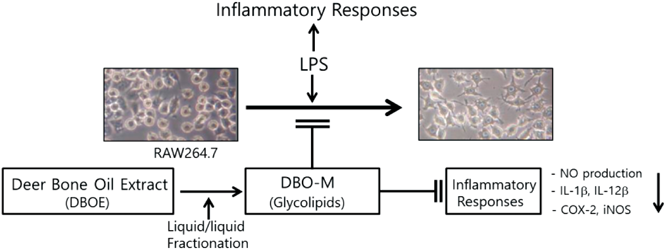 PDF形式でダウンロード (738K) HTML形式で全画面表示
PDF形式でダウンロード (738K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 601-610
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (1957K) HTML形式で全画面表示
PDF形式でダウンロード (1957K) HTML形式で全画面表示
Notes
-
2016 年 39 巻 4 号 p. 611-614
発行日: 2016/04/01
公開日: 2016/04/01
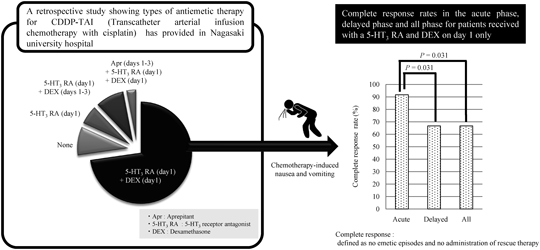 PDF形式でダウンロード (343K) HTML形式で全画面表示
PDF形式でダウンロード (343K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 615-619
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (410K) HTML形式で全画面表示
PDF形式でダウンロード (410K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 620-624
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (424K) HTML形式で全画面表示
PDF形式でダウンロード (424K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 625-630
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/28 PDF形式でダウンロード (592K) HTML形式で全画面表示
PDF形式でダウンロード (592K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 631-635
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/09 PDF形式でダウンロード (1332K) HTML形式で全画面表示
PDF形式でダウンロード (1332K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 636-640
発行日: 2016/04/01
公開日: 2016/04/01
[早期公開] 公開日: 2016/01/26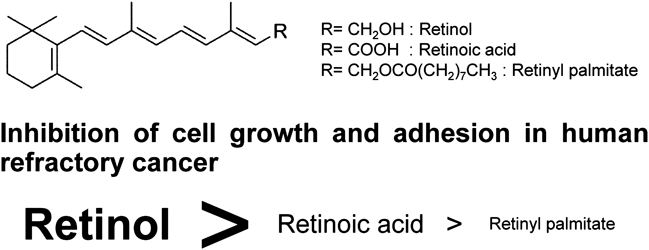 PDF形式でダウンロード (671K) HTML形式で全画面表示
PDF形式でダウンロード (671K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 641-647
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (1332K) HTML形式で全画面表示
PDF形式でダウンロード (1332K) HTML形式で全画面表示 -
2016 年 39 巻 4 号 p. 648-651
発行日: 2016/04/01
公開日: 2016/04/01
 PDF形式でダウンロード (287K) HTML形式で全画面表示
PDF形式でダウンロード (287K) HTML形式で全画面表示
Errata
-
2016 年 39 巻 4 号 p. 652
発行日: 2016/04/01
公開日: 2016/04/01
PDF形式でダウンロード (72K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|