Abstract
Melanoma is highly malignant, and generally exhibits radioresistance, responding poorly to radiation therapy. We previously reported that activation of P2X7, P2Y6, and P2Y12 receptors is involved in the DNA damage response after γ-irradiation of human lung adenocarcinoma A549 cells. However, it is not clear whether these receptors are also involved in the case of melanoma cells, although P2X7 receptor is highly expressed in various cancers, including melanoma. Here, we show that P2X7 receptor antagonist enhances radiation-induced cytotoxicity in B16 melanoma cells in vitro and in vivo. We confirmed that these cells express P2X7 receptor mRNA and exhibit P2X7 receptor-mediated activities, such as ATP-induced pore formation and cytotoxicity. We further examined the radiosensitizing effect of P2X7 receptor antagonist Brilliant Blue G (BBG) in vitro by colony formation assay of B16 cells. γ-Irradiation dose-dependently reduced cell survival, and pretreatment with BBG enhanced the radiation-induced cytotoxicity. BBG pretreatment also decreased the number of DNA repair foci in nuclei, supporting involvement of P2X7 receptor in the DNA damage response. Finally, we investigated the radiosensitizing effect of BBG on B16 melanoma cells inoculated into the hind footpad of C57BL/6 mice. Neither 1 Gy γ-irradiation alone nor BBG alone suppressed the increase of tumor volume, but the combination of irradiation and BBG significantly suppressed tumor growth. Our results suggest that P2X7 receptor antagonist BBG has a radiosensitizing effect in melanoma in vitro and in vivo. BBG, which is used as a food coloring agent, appears to be a promising candidate as a radiosensitizer.
Melanoma is a cancer of melanocytes, which are melanin-secretory cells of the skin. It is highly metastatic and has high proliferative potential, and consequently it has a poor prognosis unless diagnosed at an early stage and removed surgically. More advanced melanoma is considered a suitable target for radiation therapy to improve patients’ QOL, but melanoma has a poor therapeutic response and is radioresistant.1)
It is considered that the biological effect by γ-irradiation is mediated by reactive oxygen species (ROS), which cause DNA damage. However, the intracellular biochemical processes that occur after irradiation have not been established in detail. Recently, the bystander effect has been reported as a novel mechanism of radiation-induced biological activity.2) This effect is mediated by soluble factors released from cells in response to radiation, such as ROS, nitric oxide or cytokines.2) We have also shown that irradiated cells release nucleotides, such as ATP, ADP, uridine 5′-triphosphate (UTP) or uridine 5′-diphosphate (UDP),3,4) and that release of these nucleotides into the extracellular environment mediates cellular responses to ionizing irradiation via activation of P2 receptors.3,5)
Extracellular nucleotides (ATP, ADP, UTP, and UDP) are well known as intercellular signaling molecules.6,7) Though ATP exists at millimolar order of concentration in cells, extracellular ATP concentration is usually kept low (nanomolar order) by the action of ecto-nucleotidases. However, ATP and UTP are released from cells in response to various stimuli through maxi-anion channels, hemichannels, and anion transporters, or by exocytosis.4,8–14) These extracellular nucleotides activate P2 receptors on the cell membrane, initiating paracrine/autocrine signaling. P2 receptors are classified into two major subfamilies, ionotropic P2X1–7 receptors and metabotropic P2Y1–14 receptors, and their activation regulates many physiological functions.6,7)
One of the P2-mediated cellular responses activated by ATP is DNA repair. We have reported that γ-irradiation induces ATP release from mouse melanoma B16 cells, human keratinocyte HaCaT cells and human lung cancer A549 cells.4,15,16) P2X7 receptor-mediated connexin-hemichannel opening is involved in ATP release from B16 melanoma in response to γ-irradiation.4,11) Further, P2X7-dependent ATP release and subsequent activation of P2Y receptors, including P2Y6 and P2Y12 receptors, by released nucleotides after irradiation play important roles in DNA repair and in epidermal growth factor (EGF) receptor translocation to the nucleus, which are involved in recovery of irradiated A549 cells from cell damage.5,16,17)
We have also reported that P2Y receptor ligands enhance cell recovery and P2Y antagonists enhance cell death of irradiated A549 cells.5) Thus, it is considered that P2X7, P2Y6, and P2Y12 receptor antagonists enhance radiation-induced cell damage in irradiated cells, i.e., they have a radiosensitizing effect. However, the effect of these antagonists on melanoma has not yet been examined.
Since radiosensitization may occur not only in cancer cells, but also in normal cells expressing P2 receptors, we should select a target receptor that is highly expressed in cancer cells in order to minimize the damage to normal cells. Among the above P2 receptors, it is reported that P2X7 receptor is highly expressed in several types of cancer cells.18–22) Furthermore, involvement of P2X7 receptor in tumor growth, as well as angiogenesis of breast cancer, has been reported.18) The mechanism of P2X7 receptor-mediated tumor growth may involve production of interleukin (IL)-6 and substance P.19,20) We have previously reported that melanoma growth in vivo was inhibited by intraperitoneal (i.p.) administration of a P2X7 antagonist, oxidized ATP.23)
The main effect of exposure to radiation is DNA damage, including double-strand breaks (DSBs), and cancer cells that recover from DNA damage acquire radiation resistance.24) The DNA damage repair response is initiated within 1 h by phosphorylation of ataxia telangiectasia mutated (ATM), phosphorylated histone variant H2AX (γH2AX) focus formation, and p53-binding protein 1 (53BP1) accumulation.25–27) Although we showed that activation of P2X7 receptor is involved in the repair of DNA damage in A549 cells,16) the involvement of P2X7 receptor in DNA damage response of melanoma cells has not yet been investigated. Therefore, in this study, we investigated the radiosensitizing effect of P2X7 receptor antagonists in melanoma. We focused especially on an antagonist of P2X7 receptor Brilliant Blue G (BBG),28) which also has anti-inflammatory activity in vivo and is expected to have low toxicity,29) since it is already used as a coloring agent.29) We show here that BBG suppresses the DNA damage response of B16 melanoma cells and acts as a radiosensitizer both in vitro and in vivo.
MATERIALS AND METHODS
Reagents and AntibodiesDulbecco’s modified Eagle’s medium (DMEM) was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Gibco® fetal bovine serum (FBS) was purchased from Thermo Fisher Scientific (U.S.A). The primary antibodies used were ATM (phosphoS1981) antibody (Abcam, U.K.), 53BP1 antibody rabbit polyclonal (Novus, U.S.A.), Phospho-Histone H2AX (Ser139) rabbit monoclonal antibody (Cell Signaling Technology, U.S.A.), and purified mouse anti-H2AX phosphorylated (Ser139) antibody (Bio Legend, U.S.A.). The secondary antibodies used were Alexa Fluor 594 goat anti-mouse immunoglobulin G (IgG) (Invitrogen, U.S.A.), goat anti-rabbit IgG-fluorescein isothiocyanate (FITC) (Sigma-Aldrich, U.S.A.), and anti-rabbit IgG horseradish peroxidase (HRP)-linked antibody (Cell Signaling Technology).
Antagonists and InhibitorsA438079 (Tocris BioScience, England) is a selective antagonist of P2X7 receptor. Adenosine 5′-triphosphate disodium salt hydrate (ATP) (Sigma-Aldrich) is a ligand of P2 receptor, and we used ATP as an agonist of P2X7 receptor. BBG (Sigma-Aldrich) is an antagonist of P2X7 receptor. MRS2578 (Tocris BioScience) is a selective antagonist of P2Y6 receptor. Clopidogrel (Tocris BioScience, Wako) is a selective antagonist of P2Y12 receptor. ATM kinase inhibitor (KU55933) was purchased from Wako.
Cell CultureMouse melanoma B16 cells were grown in DMEM supplemented with 10% FBS, penicillin (100 units/mL) and streptomycin (100 µg/mL) in a humidified atmosphere of 5% CO2 in air at 37°C.
IrradiationB16 cells were irradiated with γ-rays from a Gammacell 40 (137Cs source) (Nordin International, Inc.; 0.80 Gy/min) at room temperature for an indicated time. After irradiation, the cells were incubated in a humidified atmosphere of 5% CO2 in air at 37°C.
Immunofluorescence StainingB16 cells (7.0×104 cells/mL) were seeded in a 40 mm dish and incubated for 24 h in a 5% CO2 atmosphere at 37°C, followed by exposure to various doses of γ-rays (0.5, 1.0, 2.0, 4.0, or 8.0 Gy) for indicated times (0.5, 1, 2, 3, 6, or 24 h). B16 cells were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature and permeabilized in 0.1% Triton X-100 for 5 min on ice. After incubation in blocking buffer (10% FBS in PBS) for 1 h, the fixed cells were incubated with primary antibody (γH2AX 1 : 200, 53BP1 1 : 200, ATM 1 : 1000) for 24 h at 4°C and with secondary antibody (1 : 200) for 1 h. Counterstaining with Hoechst 33258 (10 µg/mL) was used to verify the location and integrity of nuclei. Fluorescence images (magnification: ×600) were obtained with a laser scanning confocal microscope (FV1000 IX81; Olympus, Japan).
RT-PCRTotal RNA was isolated from B16 cells using a ReliaPrep™ RNA Cell Miniprep System (Promega, U.S.A.). The first-strand cDNA was synthesized from 0.5 µg of total RNA with Prime Script Reverse Transcriptase (TaKaRa Bio, Japan). The primers used for amplification were designed based on the cDNA sequences of mouse P2 receptors.30) PCR was performed by incubating each cDNA sample with appropriate primers (0.5 µM each), deoxyribonucleotide triphosphate (dNTP) mix (0.2 mM each) and Blend Taq polymerase (Toyobo, Japan) (1.25 U each). Amplification was carried out for 35 cycles (95°C for 1 min, annealing at 72°C for 2 min), followed by extension at 72°C for 10 min. The products were then subjected to 2% agarose gel electrophoresis. The bands were stained with ethidium bromide and photographed.
Analysis of Pore FormationCells were adjusted to RPMI1640-based buffer containing 102 mM NaCl, 5 mM KCl, 0.4 mM CaCl2, 0.4 mM MgSO4, 23.8 mM NaHCO3, 5.6 mM Na2HPO4, 11.1 mM glucose, and 10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)–NaOH (pH 7.4)31) at 1.0×105 cells/mL and incubated at 37°C with various concentrations of ATP and 25 µM ethidium bromide (EtBr). After incubation, each sample was analyzed using a flow cytometer (FACS Caliber cytometer, Becton, Dickinson and Co., U.K.) with laser excitation at 488 nm. Ethidium fluorescence was examined using an FL-2 filter.
Quantification of Lactate Dehydrogenase (LDH) ReleaseRelease of LDH into the cell culture supernatant was quantified with a Cytotoxicity Detection Kit (Roche Applied Science), according to the supplied instructions. B16 cells (7.0×104 cells/mL) were seeded in a 96-well plate and incubated for 24 h in a 5% CO2 atmosphere at 37°C. Cells were incubated with ATP in RPMI1640-based buffer or phenol red-free DMEM (Gibco) containing 10% FBS for 6 or 24 h at 37°C. At the end of incubation, 10 µL aliquots of culture supernatants were collected and the LDH content was measured. LDH release was expressed as a percentage of the total content determined by lysing an equal amount of cells with 1% Triton X-100. After 30 min, the absorbance at 450 nm was measured.
Colony Formation AssayB16 cells (7.0×104 cells/mL) were seeded in a 40 mm dish and incubated for 24 h in a 5% CO2 atmosphere at 37°C, followed by exposure to γ-rays (1.0, 2.0, 4.0, 8.0, or 16 Gy) for 24 h. Then, 1.0×103 cells were seeded in a 100 mm dish. After incubation for 1 week, the cells were stained with 0.5% crystal violet. Colonies containing more than 50 cells were counted. The average was obtained for each sample. The results were normalized with respect to corresponding non-irradiated cells, and the survival fraction was calculated. Antagonists were added to the medium at 30 min before irradiation.
AnimalsPathogen-free male C57BL/6 mice were purchased from Sankyo Labo Service (Tokyo, Japan) and used at 5 weeks of age. They were housed in plastic cages with paper chip bedding and bred in rooms kept at a temperature of 23±2°C and a relative humidity of 55±10% under a 12 h light–dark cycle. They were allowed free access to tap water and experimental normal diet, CE-2 (CLEA Co., Japan). The mice were treated and handled according to the Guide Principles for the Care and Use Laboratory Animals of the Japanese Pharmacological Society and the protocol was approved by Tokyo University of Science’s Institutional Animal Care and Use Committee (permission number S16003).
Transplantation ModelB16 melanoma (2.0×105 cells) was injected into the right hind footpad of 5-week-old male C57BL/6 mice. To examine the effect of P2X7 receptor antagonist on melanoma growth, 100 µL of PBS or 15 mM BBG was intraperitoneally administered to B16 melanoma-bearing mice (21.26±1.068 g) on the 11th, 14th, 18th, and 21st days after transplantation. Mice were irradiated with 1.0 Gy of γ-rays at 2 h after treatment with BBG on the 11th, 14th, 18th, and 21st days after transplantation. The size of the solid tumor was measured with a caliper twice a week for 24 d, and the tumor volume (V) was calculated according to the following equation;
StatisticsResults are expressed as the mean±standard error (S.E.). The statistical significance of differences between control and other groups was calculated using Dunnett’s test. Multiple groups were compared using ANOVA followed by pairwise comparisons with Bonferroni’s post hoc analysis. Calculations were done with the Instat version 3.0 statistical software package (Graph Pad Software). The criterion of significance was p<0.05.
RESULTS
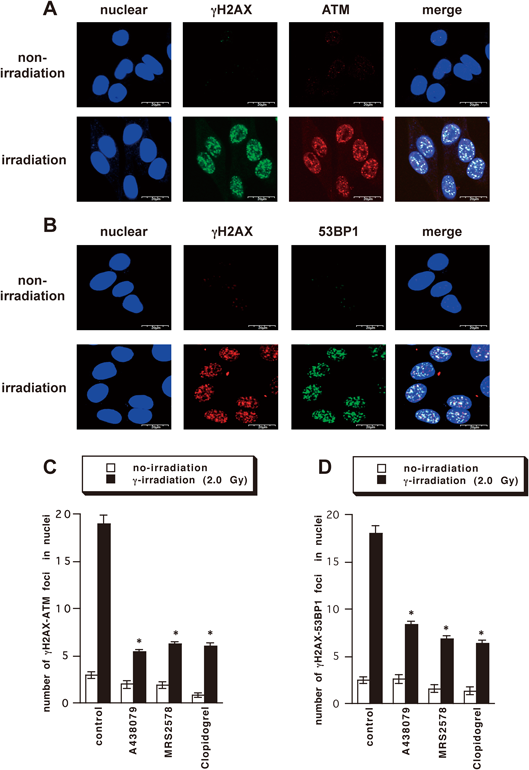
DNA Damage Caused by γ-Irradiation and Involvement of Purinergic ReceptorsFirst, we examined formation of γH2AX foci after γ-irradiation in B16 melanoma cells by immunofluorescence staining. γH2AX focus formation reached a maximum at 0.5–3 h after irradiation (Figs. 1A, B) and increased in a dose-dependent manner (0.5–8.0 Gy) (Fig. 1C). We have reported that extracellular ATP, which is released by irradiation via P2X7 receptor, facilitates formation of DNA repair foci at γ-irradiation-induced DNA damage sites via activation of P2Y6 or P2Y12 receptors in A549 cells.5,16) Therefore, we evaluated the effect of P2X7, P2Y6, and P2Y12 receptor antagonists on activation of ATM and accumulation of 53BP1 at DNA damage sites in irradiated B16 melanoma (Fig. 2). As shown in Fig. 2C, the number of co-stained activated ATM and γH2AX foci in B16 melanoma was decreased by pretreatment with P2X7 receptor antagonist A438079, P2Y6 receptor antagonist MRS2578, and P2Y12 receptor antagonist clopidogrel. Moreover, the number of co-stained 53BP1 and γH2AX foci was also reduced by pretreatment with A438079, MRS2578, and Clopidogrel (Fig. 2D). These results indicate that P2X7, P2Y6, and P2Y12 receptors are all involved in DNA damage responses in B16 melanoma.
Evaluation of P2X7 Receptor Activity in B16 Mouse Melanoma CellsP2X7 receptor expression is increased in many cancer tissues, including thyroid cancer and melanoma.32) We have previously demonstrated expression and activity of P2X7 receptor in B16 melanoma.4,23) Here, we examined mRNA expression of P2 receptor subtypes in B16 melanoma cells by RT-PCR. As shown in Fig. 3A, various P2 receptor mRNAs were observed. In particular, P2X7 receptor was expressed in B16 melanoma cells. To examine the activity of P2X7 receptor in B16 melanoma cells, we evaluated P2X7 receptor-dependent pore formation in terms of EtBr uptake (Figs. 3B, C), because activation of P2X7 receptor causes formation of non-selective pores allowing passage of molecules of up to 900 kDa. Pore formation was observed at 10 min after addition of 1.0–3.0 mM ATP, and the ATP-induced pore formation was suppressed by pretreatment with A438079 (Figs. 3B, C).
Since strong and sustained activation of P2X7 receptor induces cell death in various types of cells, such as immune cells,33,34) we examined the release of LDH induced by P2X7 agonist ATP (0.5–3.0 mM). In B16 melanoma cells incubated in buffer not containing FBS according to our previous study,35) the release of LDH was increased by a high concentration of ATP (1.0 or 3.0 mM), and pretreatment with P2X7 antagonist BBG blocked this release (Fig. 3D). Thus, we confirmed that B16 cells highly express functional P2X7 receptor.
Contribution of P2X7 Receptor to Cell Recovery after γ-IrradiationWe next investigated whether P2X7 receptor plays a role in cell recovery after γ-irradiation. We examined the survival fraction 1 week after irradiation (1.0–16 Gy of γ-rays) by colony formation assay. γ-Irradiation-induced cell death increased in a dose-dependent manner (Fig. 4A). We examined the effects of two P2X7 receptor antagonists, BBG (10–100 µM) and A438079. Pretreatment with BBG significantly enhanced the decrease of survival fraction caused by 2.0 Gy γ-irradiation in a dose-dependent manner (Fig. 4B). Pre-treatment with A438079 (a selective P2X7 receptor antagonist) also enhanced the decrease of survival fraction (Fig. 4C).
On the other hand, treatment with 1.0 mM ATP, which activates P2X7 receptor tended to attenuate the decrease of survival fraction caused by 2.0 Gy irradiation (Fig. 4D). We confirmed that ATP-induced cell death was not observed in B16 cells incubated in the medium containing 10% FBS at either 6 or 24 h after addition of 1.0 mM ATP (Figs. 5A, B), which are the same conditions used for colony formation assay. The presence of various growth factors in serum might attenuate the induction of cell death through activation of P2X7 receptor, though the mechanism is not established. These results indicate that P2X7 receptor plays an important role in cell survival after irradiation-induced DNA damage.
Involvement of P2X7 Receptor in DNA Damage RepairWe examined the effects of BBG on γ-irradiation-induced DNA repair. As shown in Figs. 6A and B, the γ-irradiation-induced increase of co-localization of activated ATM or 53BP1 with γH2AX foci was significantly suppressed by pretreatment with BBG, suggesting that P2X7 antagonist BBG inhibited DNA damage responses after γ-irradiation. We confirmed that treatment with exogenous ATP did not increase DNA repair foci in non-irradiated cells (Figs. 6A, C). These results indicate that ATP released from cells after γ-irradiation triggered DNA repair responses via activation of P2 receptors in irradiated B16 melanoma cells. We confirmed the importance of ATM kinase in DNA repair and cell survival of irradiated cells by showing that an inhibitor of ATM kinase significantly enhanced the decrease of survival fraction caused by 2.0 Gy γ-irradiation (Fig. 6C).
Enhanced Suppression of Melanoma Growth by Combination of γ-Irradiation with BBGThe above results indicate that P2X7 receptor contributes significantly to recovery of irradiated cells from potentially lethal cell damage, providing a new insight in the mechanism of radiation-induced cellular responses in B16 melanoma cells.
To see whether this might have implications in vivo, we finally examined the radiosensitizing effect of BBG on growth of B16 melanoma cells transplanted into footpad of C57BL/6 mice. The mice were treated with BBG (1.28 mg/head i.p.), and then given whole-body γ-irradiation (1.0 Gy) at the 11th, 14th, 18th, and 21st days after transplantation (total, 4.0 Gy irradiation). We measured tumor volume at the 11th, 14th, 18th, 21st, and 24th days after transplantation. As shown in Fig. 7, radiation therapy alone did not suppress melanoma growth in vivo, and treatment with BBG only slightly suppressed the melanoma growth. However, irradiation of mice pretreated with BBG causes significant suppression of melanoma growth compared with the irradiation alone group, indicating that BBG acts as a radiosensitizer not only in vitro, but also in vivo.
DISCUSSION
Expression of the P2X7 receptor is observed in B16 melanoma cells, and it has been proposed that this increased expression can serve as a biomarker for cancer.36–38) Considering these results, together with our previous work on the role of P2X7 receptor in promoting radiation-induced DNA repair,5,16) we hypothesized that antagonists of the P2X7 receptor might serve as effective radiosensitizers of melanoma with high specificity over adjacent normal cells.
We previously showed that P2X7 receptor-dependent ATP release occurs at 1–10 min after irradiation of B16 melanoma cells,4,11) and the released ATP induces formation of γH2AX foci, a marker of DNA damage and DNA repair, peaking at 30 min after irradiation. Though the reason why γH2AX formation was peaked twice at 1 and 3 h after irradiation in B16 cells, we speculate that DNA damage response might be induced by both first acute response within 1 h and second signaling caused by intracellular/intercellular signaling to amplify DNA repair. Here we confirmed that similar irradiation-induced changes occurred dose-dependently in B16 melanoma cells. Further, we found that P2X7 receptor antagonists suppressed the formation of γH2AX foci, as well as activation of ATM and accumulation of 53BP1, which are also DNA repair responses. These results confirmed that P2X7 receptor is involved in repair of radiation-induced DNA damage in B16 mouse melanoma cells.
In this study, we focused mainly on BBG as a P2X7 antagonist,28) because it has been used as an artificial color in foods for a long time.29,39) We examined its cytotoxicity by means of WST-1 assay and found that proliferation of B16 melanoma cells was not suppressed by 10–100 µM BBG, although proliferation was somewhat decreased by 500–1000 µM BBG (data not shown). Thus, we considered that BBG would have little cytotoxicity towards B16 cells at up to 100 µM concentration. We also confirmed that pretreatment with 50 µM BBG was sufficient to suppress ATP-induced activation of P2X7 receptor in B16 cells.
γ-Irradiation dose-dependently induced reproductive cell death of B16 melanoma cells in the range from 1.0 to 16 Gy, and its action was enhanced by pretreatment with 10–100 µM BBG, confirming the expected radiosensitizing effect of the antagonist. We also found that 1.0 mM ATP, a P2X7 receptor agonist, attenuated reproductive cell death, supporting the idea that activation of P2X7 receptor facilitates cell recovery. This is consistent with a previous report that ATP protected lymphocytes from radiation damage.40) The other P2X7 antagonist examined here, A438079, suppressed DNA damage responses similarly to BBG.
It was recently reported that ATP enhances cell death caused by γ-irradiation in glioma M059J cells,41) which appears to conflict with our findings. However, DNA protein kinase, a DNA damage repair enzyme, is defective in M059J cells, and this is likely to impede DNA damage repair.42) This may explain why P2X7 receptor activation with ATP failed to facilitate DNA damage responses in irradiated M059J cells. Furthermore, in B16 melanoma, P2X7-dependent cell death (measured in terms of LDH release) was not observed in the presence of serum, suggesting that the role of P2X7 receptor in the cellular response to irradiation might be different between glioma and melanoma.
It has been reported that BBG inhibited growth/metastasis of glioma cells.43) However, there has been no report on the combined use of BBG with γ-irradiation in vivo. We therefore examined the radiosensitizing effect of BBG in vivo in melanoma-transplanted mice. Melanoma growth was not suppressed by 1.0 Gy γ-irradiation alone, and was only slightly suppressed by BBG alone. However, the combination of 1.0 Gy γ-irradiation with BBG treatment did suppress tumor growth, suggesting that BBG can serve as a radiosensitizer of melanoma in vivo.
Cancer cells express many P2 receptors, such as P2X5, P2X7, P2Y1, P2Y2, and P2Y11, and activation of these receptors causes a variety of cellular responses in cancer cells.44) Some purine receptors have been reported to be involved in tumor growth,45) and it was found that P2X7 receptor inhibitors block tumor growth.18) These findings are consistent with the idea that BBG suppresses tumor growth/metastasis and enhances the efficacy of radiotherapy by blocking activation of P2X7 receptor.
It is also well known that P2X7 receptor in immune cells significantly contributes to inflammation via induction of pro-inflammatory cytokines, and BBG was shown to suppress secretion of inflammatory cytokines in rat.46) The P2X7 receptor is also involved in nociception.47) Thus, BBG might have multiple therapeutic effects.
In conclusion, we found that P2X7 antagonist BBG enhances radiation damage in melanoma by suppressing DNA damage repair response, thereby acting as a radiosensitizer in vitro and in vivo. Various molecules have been investigated as radiosensitizers, including gold nanoparticles and inhibitors of DNA repair enzymes,48,49) but this is the first evidence that P2X7 receptor antagonists can serve as radiosensitizers. We suggest that BBG is a promising candidate as a radiosensitizer for radiation therapy of melanoma, since it appears to have multiple activities, suppressing not only tumor growth, but also metastasis, inflammation and pain. It would also be interesting to investigate the effect of BBG on other types of cancer.
Acknowledgment
Parts of this work were supported by Fukushima Prefecture (Grants for pursuing the goal of being a world-class center of medically related industries).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Satyamoorthy K, Chehab NH, Waterman MJ, Lien MC, El-Deiry WS, Herlyn M, Halazonetis TD. Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ., 11, 467–474 (2000).
- 2) Hamada N, Maeda M, Otsuka K, Tomita M. Signaling pathways underpinning the manifestations of ionizing radiation-induced bystander effects. Curr. Mol. Pharmacol., 4, 79–95 (2011).
- 3) Tsukimoto M. Purinergic signaling is a novel mechanism of the cellular response to ionizing radiation. Biol. Pharm. Bull., 38, 951–959 (2015).
- 4) Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, Kitani H, Kojima S. γ-Irradiation induces P2X7 receptor-dependent ATP release from B16 melanoma cells. Biochim. Biophys. Acta, 1800, 40–46 (2010).
- 5) Ide S, Nishimaki N, Tsukimoto M, Kojima S. Purine receptor P2Y6 mediates cellular response to gamma-ray-induced DNA damage. J. Toxicol. Sci., 39, 15–23 (2014).
- 6) Burnstock G. Purinergic signalling: past, present and future. Braz. J. Med. Biol. Res., 42, 3–8 (2009).
- 7) Burnstock G. Purinergic signalling. Br. J. Pharmacol., 147 (Suppl.), S172–S181 (2006).
- 8) Baroja-Mazo A, Barberà-Cremades M, Pelegrín P. The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta, 1828, 79–93 (2013).
- 9) Hisadome K, Koyama T, Kimura C, Droogmans G, Ito Y, Oike M. Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J. Gen. Physiol., 119, 511–520 (2002).
- 10) Liu H-T, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res., 18, 558–565 (2008).
- 11) Ohshima Y, Tsukimoto M, Harada H, Kojima S. Involvement of connexin43 hemichannel in ATP release after γ-irradiation. J. Radiat. Res., 53, 551–557 (2012).
- 12) Pangršič T, Potokar M, Stenovec M, Kreft M, Fabbretti E, Nistri A, Pryazhnikov E, Khiroug L, Giniatullin R, Zorec R. Exocytotic release of ATP from cultured astrocytes. J. Biol. Chem., 282, 28749–28758 (2007).
- 13) Sabirov RZ, Dutta AK, Okada Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J. Gen. Physiol., 118, 251–266 (2001).
- 14) Stout CE, Costantin JL, Naus CCG, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem., 277, 10482–10488 (2002).
- 15) Tsukimoto M, Homma T, Ohshima Y, Kojima S. Involvement of purinergic signaling in cellular response to γ-radiation. Radiat. Res., 173, 298–309 (2010).
- 16) Nishimaki N, Tsukimoto M, Kitami A, Kojima S. Autocrine regulation of γ-irradiation-induced DNA damage response via extracellular nucleotides-mediated activation of P2Y6 and P2Y12 receptors. DNA Repair (Amst.), 11, 657–665 (2012).
- 17) Tamaishi N, Tsukimoto M, Kitami A, Kojima S. P2Y6 receptors and ADAM17 mediate low-dose gamma-ray-induced focus formation (activation) of EGF receptor. Radiat. Res., 175, 193–200 (2011).
- 18) Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res., 72, 2957–2969 (2012).
- 19) Solini A, Cuccato S, Ferrari D, Santini E, Gulinelli S, Callegari MG, Dardano A, Faviana P, Madec S, Di Virgilio F, Monzani F. Increased P2X7 receptor expression and function in thyroid papillary cancer: a new potential marker of the disease? Endocrinology, 149, 389–396 (2008).
- 20) Raffaghello L, Chiozzi P, Falzoni S, Di Virgilio F, Pistoia V. The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res., 66, 907–914 (2006).
- 21) Greig AVH, Linge C, Healy V, Lim P, Clayton E, Rustin MHA, McGrouther DA, Burnstock G. Expression of purinergic receptors in non-melanoma skin cancers and their functional roles in A431 cells. J. Invest. Dermatol., 121, 315–327 (2003).
- 22) Slater M, Danieletto S, Pooley M, Cheng Teh L, Gidley-Baird A, Barden JA. Differentiation between cancerous and normal hyperplastic lobules in breast lesions. Breast Cancer Res. Treat., 83, 1–10 (2004).
- 23) Hattori F, Ohshima Y, Seki S, Tsukimoto M, Sato M, Takenouchi T, Suzuki A, Takai E, Kitani H, Harada H, Kojima S. Feasibility study of B16 melanoma therapy using oxidized ATP to target purinergic receptor P2X7. Eur. J. Pharmacol., 695, 20–26 (2012).
- 24) Betz H. Importance of the phase of resistance of the organism during the application of a lethal dose of X-rays. C. R. Seances Soc. Biol. Fil., 144, 1439–1442 (1950).
- 25) Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol., 3, 113 (2013).
- 26) Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst.), 9, 1219–1228 (2010).
- 27) Ataian Y, Krebs JE. Five repair pathways in one context: chromatin modification during DNA repair. Biochem. Cell Biol., 84, 490–494 (2006).
- 28) Jiang LH, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol. Pharmacol., 58, 82–88 (2000).
- 29) Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian G-F, Goldman SA, Nedergaard M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl. Acad. Sci. U.S.A., 106, 12489–12493 (2009).
- 30) Ishimaru M, Yusuke N, Tsukimoto M, Harada H, Takenouchi T, Kitani H, Kojima S. Purinergic signaling via P2Y receptors up-mediates IL-6 production by liver macrophages/Kupffer cells. J. Toxicol. Sci., 39, 413–423 (2014).
- 31) Tsukimoto M, Maehata M, Harada H, Ikari A, Takagi K, Degawa M. P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J. Immunol., 177, 2842–2850 (2006).
- 32) Deli T, Varga N, Ádám A, Kenessey I, Rásó E, Puskás LG, Tóvári J, Fodor J, Fehér M, Szigeti GP, Csernoch L, Tímár J. Functional genomics of calcium channels in human melanoma cells. Int. J. Cancer, 121, 55–65 (2007).
- 33) Burnstock G, Boeynaems J-M. Purinergic signalling and immune cells. Purinergic Signal., 10, 529–564 (2014).
- 34) Toki Y, Takenouchi T, Harada H, Tanuma S, Kitani H, Kojima S, Tsukimoto M. Extracellular ATP induces P2X7 receptor activation in mouse Kupffer cells, leading to release of IL-1β, HMGB1, and PGE2, decreased MHC class I expression and necrotic cell death. Biochem. Biophys. Res. Commun., 458, 771–776 (2015).
- 35) Kawano A, Tsukimoto M, Noguchi T, Hotta N, Harada H, Takenouchi T, Kitani H, Kojima S. Involvement of P2X4 receptor in P2X7 receptor-dependent cell death of mouse macrophages. Biochem. Biophys. Res. Commun., 419, 374–380 (2012).
- 36) Slater M, Scolyer RA, Gidley-Baird A, Thompson JF, Barden JA. Increased expression of apoptotic markers in melanoma. Melanoma Res., 13, 137–145 (2003).
- 37) Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science, 272, 735–738 (1996).
- 38) White N, Butler PEM, Burnstock G. Human melanomas express functional P2 X(7) receptors. Cell Tissue Res., 321, 411–418 (2005).
- 39) Díaz-Hernández M, Díez-Zaera M, Sánchez-Nogueiro J, Gómez-Villafuertes R, Canals JM, Alberch J, Miras-Portugal MT, Lucas JJ. Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J., 23, 1893–1906 (2009).
- 40) Swennen ELR, Dagnelie PC, Van den Beucken T, Bast A. Radioprotective effects of ATP in human blood ex vivo. Biochem. Biophys. Res. Commun., 367, 383–387 (2008).
- 41) Gehring MP, Pereira TCB, Zanin RF, Borges MC, Braga Filho A, Battastini AMO, Bogo MR, Lenz G, Campos MM, Morrone FB. P2X7 receptor activation leads to increased cell death in a radiosensitive human glioma cell line. Purinergic Signal., 8, 729–739 (2012).
- 42) Virsik-Köpp P, Rave-Fränk M, Hofman-Hüther H, Schmidberger H. Role of DNA-PK in the process of aberration formation as studied in irradiated human glioblastoma cell lines M059K and M059J. Int. J. Radiat. Biol., 79, 61–68 (2003).
- 43) Ryu JK, Jantaratnotai N, Serrano-Perez MC, McGeer PL, McLarnon JG. Block of purinergic P2X7R inhibits tumor growth in a C6 glioma brain tumor animal model. J. Neuropathol. Exp. Neurol., 70, 13–22 (2011).
- 44) White N, Burnstock G. P2 receptors and cancer. Trends Pharmacol. Sci., 27, 211–217 (2006).
- 45) Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal., 9, 491–540 (2013).
- 46) Gourine AV, Poputnikov DM, Zhernosek N, Melenchuk EV, Gerstberger R, Spyer KM, Gourine VN. P2 receptor blockade attenuates fever and cytokine responses induced by lipopolysaccharide in rats. Br. J. Pharmacol., 146, 139–145 (2005).
- 47) Burnstock G. Purinergic mechanisms and pain. Adv. Pharmacol., 75, 91–137 (2016).
- 48) Jain S, Hirst DG, O’Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Br. J. Radiol., 85, 101–113 (2012).
- 49) Cooper DR, Bekah D, Nadeau JL. Gold nanoparticles and their alternatives for radiation therapy enhancement. Front. Chem., 2, 86 (2014).