MATERIALS AND METHODS
ChemicalsTelmisartan and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Paclitaxel and fluvastatin were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Irbesartan, which was used as an internal standard (IS) for the determination of the fluvastatin metabolites, and candesartan were obtained from Astatech (Bristol, PA, U.S.A.). Docetaxel trihydrate, which was used as an IS for the determination of 6α-hydroxypaclitaxel, and 6α-hydroxypaclitaxel were purchased from Toronto Research Chemicals (Toronto, Canada) and Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.), respectively. Supersomes™ from insect cells that expressed recombinant human CYP2C9 (rCYP2C9)*1 (lot: 4141001), rCYP2C9*2 (lot: 5056008), rCYP2C9*3 (lot: 4203003), and control microsome (lot: 5217001), along with the 150-donor pooled human liver microsomes (pooled HLMs, lot: 38291) and monoclonal antibody for human CYP2C8 (MAB-2C8) derived from mouse were all obtained from Corning (Woburn, MA, U.S.A.). Metabolites of fluvastatin (M-2, M-3, and M-5) were kindly provided by Novartis Pharma (Tokyo, Japan). Other reagents and solvents were of either HPLC or special grades.
Estimation of the Effect of the Contribution Ratios (CRs) and the Fraction of the Activity of the CYP2C9 Mutated Allele Relative to the Wild-Type Allele (FA2C9) on Fluvastatin MetabolismEstimations of the effects of the CRs of CYP2C9 (CR2C9) and CYP3A4 (CR3A4) on the fluvastatin metabolism were determined in accordance with the method already validated,7,8,18,19) using data derived from two in vivo interaction studies.21,22) CR2C9 and CR3A4 were estimated using the following equation, which was initially proposed by Tod et al.,19) with slight modification:
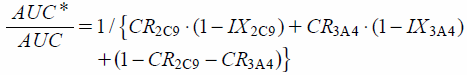 | (1) |
where
AUC* and
AUC are
AUCs in patients in the presence or absence of inhibitor, respectively.
CR2C9 and
CR3A4 represent the
CRs of CYP2C9 and CYP3A4 to the fluvastatin metabolism, respectively. The potency of the inhibitor (
IX) for CYP2C9 and CYP3A4 were
IX2C9 and
IX3A4, respectively, with the values ranging from 0 (no inhibition) to 1 (complete inhibition),
e.g., 0.5 of
IX2C9 represents that the inhibitor cause the 50% inhibition of CYP2C9 activity in clinical setting. In two
in vivo interaction studies in relation to fluvastatin metabolism,
21,22) fluconazole and itraconazole were used as inhibitors, and
IXs of those inhibitors have been reported elsewhere
7,19) so that we used those values of
IXs for the further analysis. Kantola
et al. reported that 200 mg of fluconazole, which inhibits both CYP2C9 and CYP3A4, caused a 1.84-fold increase in the fluvastatin
AUC,
21) and the
IX2C9 and
IX3A4 of fluconazole have been reported to be 0.56
19) and 0.79,
7) respectively. Kivistö
et al. reported that 100 mg of itraconazole, which is a potent inhibitor for CYP3A4, caused a 1.27-fold increase in the fluvastatin
AUC,
22) and the
IX3A4 of itraconazole was reported to be 0.95.
7) We estimated the
CR2C9 to be 0.63, while the values of
CR3A4 to be 0.13 and 0.22. Based on these findings, we used 0.18 as the mean value of the
CR3A4 in our further analyses.
The in vitro inhibition experiment was carried out using MAB-2C8 in order to estimate the CR2C8, which represents the CR of CYP2C8 to fluvastatin metabolism. In accordance with manufacturer’s instruction and a previous report,23) the incubation mixture containing the pooled HLMs (62.5 µg protein in a total incubation volume of 250 µL) in 50 mM Tris buffer (pH 7.4) was pre-incubated with 3.1 µL of MAB-2C8 (0.5 mg MAB protein/mg HLMs protein) and 6.9 µL of 25 mM Tris buffer (pH 7.5) for 20 min on ice. For the control incubation sample, pooled HLMs were pre-incubated with normal mouse immunoglobulin G (IgG) (Santa Cruz Biotechnology) to assess the nonspecific inhibition. We confirmed that MAB-2C8 completely inhibited CYP2C8 activity, whereas had negligible effect for the activity of CYP2C9 and CYP3A4 in these experimental conditions using paclitaxel and losartan as substrates of those CYP isoforms prior to this inhibition study. Fluvastatin dissolved in methanol was added to each sample (final concentration: 5 µM4)), which resulted in a 1% methanol concentration in each incubation mixture. The mixture was pre-warmed in a shaking water bath at 37°C for 3 min, and then NADPH (final concentration: 1 mM) was added to initiate the reaction. After incubation for 15 min, 800 µL of ice-cold acetonitrile with IS (200 nM irbesartan) was added to stop the reaction. All incubations were performed in triplicate. After centrifugation of all samples at 6490×g for 10 min, the supernatant was evaporated to dryness. The residue was reconstituted with 75 µL of mobile phase, and then centrifuged at 6490×g for 10 min. Subsequently, 50 µL of the supernatant was injected into the HPLC. Metabolites of fluvastatin were determined using a previously described method4) with slight modification. The mobile phase consisted of acetonitrile and 0.3% phosphoric acid (42 : 58, v/v), with the flow rate set at 1.0 mL/min. Analysis was performed using a fluorescence detector (RF-10A; Shimadzu, Kyoto, Japan) with the excitation and emission wavelengths set at 305 and 390 nm, respectively. The limits of quantification for M-2, M-3, and M-5 were 2, 70, and 1 nM, respectively. All incubations were carried out within the linear range of fluvastatin metabolism with respect to the protein concentration, incubation time and substrate concentration.
The FA2C9 was calculated using a method as described elsewhere.18,19) Briefly, various concentrations of fluvastatin were incubated with rCYP2C9*1, rCYP2C9*2, or rCYP2C9*3, and used to estimate the maximum velocity (Vmax) and Michaelis constant (Km) in relation to each metabolite formation. Protein concentration in an incubation mixture was adjusted with insect cell control microsome to yield a final concentration of 0.5 mg protein/mL. Fluvastatin concentrations were set at 0.5, 0.75, 1.0, 2.5, 5.0, 10, and 25 µM. Incubation time was set at 10 min for rCYP2C9*3 and 15 min for the others. Metabolites of fluvastatin were determined using the HPLC method described above. The Vmax and Km were estimated by the nonlinear least-squares fitting method of the GraphPad Prism software version 5.0 (GraphPad Software Inc.; San Diego, CA, U.S.A.). The intrinsic clearance (CLint) was then calculated using these parameters after correction with an intersystem extrapolation factor (ISEF), microsomal protein per gram of liver (MPPGL), and liver weight by the following equation24):
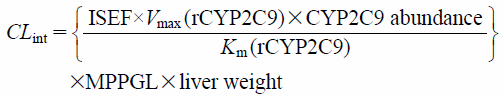 | (2) |
where
Vmax (rCYP2C9) and
Km (rCYP2C9) are the
Vmax and
Km towards fluvastatin metabolism in rCYP2C9, respectively. Since we used rCYP2C9 derived from baculovirus expression system, the
CLint was corrected with the CLISEF value of 2.64 as described by Proctor
et al.24) CYP2C9 abundance was used of 61 pmol/mg protein that was reported in a meta-analysis.
25) The values of 34 mg protein/g of liver and 1603 g were used as MPPGL and liver weight, respectively.
26,27)Assuming that there are m categories of alleles and ni alleles in each category, we estimated the FA2C9 for the various CYP2C9 genotypes by using the following equation18):
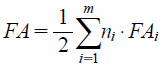 | (3) |
where
FAi is the fraction of activity on fluvastatin metabolism, corrected
CLint, of the variant CYP2C9 allele relative to the wild-type CYP2C9 allele. The reference value of the
FA2C9 in the carriers who had the homozygote wild-type CYP2C9 was equal to 1.
18)Prediction of the Increase in the AUC of Fluvastatin Caused by the CYP2C9 Genetic VariantsTo predict the increase in the AUC of fluvastatin in carriers with mutated CYP2C9 allele versus that in the patients homozygous for CYP2C9*1 at the same dosage (AUCXM/AUCEM), we used the following validated equation18,28):
 | (4) |
where XM and EM represent the parameters in the carriers with the mutated CYP2C9 allele and the wild-type CYP2C9 allele, respectively. Predicted values of the increase in the
AUC were then compared with the observed 3R, 5S-fluvastatin
AUC values
11) in carrier of CYP2C9 genetic variants. According to previous literatures,
18,19) predicted values that were 50–200% of observed ratios were considered correct.
Prediction of the Increase in the AUC of Fluvastatin in Carriers with the CYP2C9 Genetic Variants Caused by DDI Compared to That Observed in CYP2C9 Wild-Type Carriers Taking Fluvastatin AloneThe increase of the AUC of fluvastatin in the carriers of mutated CYP2C9 alleles caused by DDI (AUCXM*/AUCEM) was predicted by using the following validated equation19):
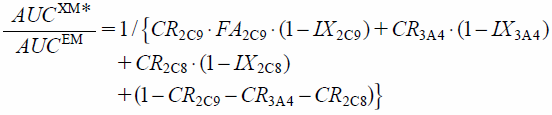 | (5) |
where
AUCXM* is the
AUC of fluvastatin in carriers with the mutated CYP2C9 allele when fluvastatin is administered with CYP inhibitors.
AUCEM is the
AUC of fluvastatin in carriers with the CYP2C9 wild-type taking fluvastatin alone.
Owing to the luck of information about in vivo interaction studies that telmisartan and candesartan were used as CYP inhibitors, we estimated the IXs of these drugs were estimated using in vitro data and the following equation7,8,19):
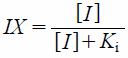 | (6) |
where [
I] is the concentration of the inhibitor in the liver, which is usually the unbound concentration of inhibitor. However, since telmisartan is a substrate of the organic anion transporter (OATP) 1B3,
29) there is a high accumulation in the human liver after oral administration. This indicates that predictions of the
IXs for telmisartan when using the unbound concentration would cause a false negative result, as predictions based on the unbound concentration of the inhibitor assume that the inhibitor distributes into the liver solely by passive diffusion. Candesartan does not accumulate in the human liver, which indicates that its distribution in the liver occurs mainly by passive diffusion. Therefore, in order to avoid any false negative results, we calculated the
IXs of telmisartan with the maximum total hepatic inlet concentration ([
I]
in) and candesartan with the maximum total concentration in the blood (
Cmax,blood).
30) [
I]
in was calculated by the following equation:
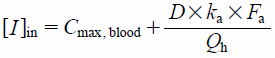 | (7) |
where
Cmax,blood and
D represent the maximum total concentration in the blood and the dosage of telmisartan, respectively.
Qh is the hepatic blood flow,
ka is the first-order absorption rate constant, and
Fa is the fraction of the oral dose absorbed. Values of 1610 mL/min, 0.1 min
−1, and 1.0 were used for the
Qh,
ka, and
Fa, respectively.
30) The unbound concentrations of [
I]
in ([
I]
in,u) were also calculated by multiplying [
I]
in and the plasma protein unbound fraction of telmisartan, 0.005.
31)The maximum concentration of the candesartan in the plasma after multiple oral administrations at the maximum clinical dose of 16 mg/d has been reported to be 0.264 µM.32) Since candesartan does not appear to distribute to blood cells,33) we used 0.55 as the smallest blood to plasma concentration ratio (Rb). Thus, the highest Cmax,blood calculated was 0.145 µM. A previous study has examined the maximum concentrations of telmisartan that occurred in the plasma when using various dosages for multiple oral administrations.34) Furthermore the Rb of telmisartan has been reported to be 0.8.35) The Cmax,blood values for the various dosages of telmisartan were calculated by multiplying the Rb and the corresponding plasma concentrations.34) Next, the [I]in and [I]in,u for the various dosages and the IX2C9 were calculated using Eqs. 6 and 7 (Supplementary Tables 1, 2).
The Ki values of telmisartan and candesartan for CYP2C9 were estimated from previous in vitro inhibition experiments.14,15) The authors reported the half maximal inhibitory concentration (IC50) values calculated from incubation experiments under the condition that the substrates concentrations equaled to those Km. Therefore, we calculated their Ki values using the following equation36):
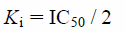 | (8) |
we determined the
Ki values of telmisartan for CYP2C9 to be 21.0
14) and 2.39 µM,
15) respectively. We then used the mean value, 11.7 µM, for our further analysis of telmisartan. The IC
50 value of candesartan for CYP2C9 has been reported to be 57.7 µM,
14) and the
Ki value for CYP2C9 was determined to be 28.9 µM.
After we carried out an in vitro inhibition experiment of paclitaxel with pooled HLMs by using telmisartan and candesartan to determine the IC50 values for CYP2C8, we then calculated the Ki values for CYP2C8 using Eq. 8. The inhibition experiment was performed according to our previous report37) with slight modification. HLMs (1.0 mg protein/mL) were incubated with inhibitors, paclitaxel, and NADPH (1 mM) in Tris–HCl buffer (50 mM, pH 7.4) in a total volume of 250 µL. Both of the inhibitor concentrations were set at 0, 5, 10, 50, 100, 500 µM, while the paclitaxel concentration was set at 7.5 µM, which was equal to the Km. After incubation for 30 min, the reaction was stopped by adding 1 mL of ice-cold acetonitrile containing 1 µg/mL of docetaxel (IS). The concentration of 6α-hydroxypaclitaxel produced in each of the incubation mixtures was determined by an HPLC-UV method that has been previously described.37) Mobile phase was delivered isocratically with 20 mM ammonium acetate buffer (pH 5.0) and acetonitrile (58 : 42, v/v) at a flow rate of 0.9 mL/min. The detection wavelength was set at 240 nm. The limit of quantification for 6α-hydroxypaclitaxel was 100 ng/mL (115 nM). All incubations were carried out within the linear range of paclitaxel metabolism with respect to the protein concentration, incubation time and substrate concentration. IC50 values were calculated using the nonlinear least-squares fitting method of the GraphPad Prism software. All incubations were performed in triplicate.
RESULTS
Estimation of the CR2C8 and FA2C9 on the Fluvastatin MetabolismThe overall metabolic rate of fluvastatin in HLMs with MAB-2C8 and normal mouse IgG were 168.7±3.6 and 186.6±4.4 pmol/mg protein/min, respectively (mean±standard deviation (S.D.), n=3). MAB-2C8 significantly inhibited the overall metabolism of fluvastatin by 10% (p=0.0054 by Student’s t-test), with the CR2C8 for the fluvastatin metabolism estimated to be 0.10.
The estimated enzyme kinetic parameters and FA2C9 values from our experiments that used rCYP2C9s are shown in Tables 1, 2, respectively. CYP2C9*2 caused an increase in the Km value for all of the fluvastatin metabolites, which resulted in a 42% decrease of the CLint compared to CYP2C9*1. CYP2C9*3 caused a decrease in the Vmax and an increase in the Km, which resulted in an 81% decrease of the CLint compared to CYP2C9*1.
Table 1. Enzyme Kinetic Parameters for the Fluvastatin Metabolism by rCYP2C9s
| Enzyme kinetic parameters | rCYP2C9*1 | rCYP2C9*2 | rCYP2C9*3 |
|---|
| Vmax (fmol/pmol P450/min)[95% CI] | 273.6 [256.6–290.7] | 271.3 [259.8–286.5] | 193.4 [169.8–216.9] |
| Km (µM) [95% CI] | 1.1 [0.87–1.4] | 1.9 [1.6–2.2] | 4.1 [2.7–5.5] |
| CLint (nL/pmol P450/min) | 248.7 | 143.7 | 47.2 |
| Corrected CLint (L/h) | 131.0 | 75.7 | 24.8 |
Each parameter was estimated by using the nonlinear least-squares method and the means of the three metabolites formation determined from triplicate incubations. 95% CI: 95% confidence interval.
Table 2. Estimates of the Fraction of the Activity of the Mutated CYP2C9 Alleles Relative to the CYP2C9 Wild-Type Allele (
FA2C9) Obtained in the Current Study and Previous Meta-Analyses
| CYP2C9 genotypes | FA2C9 |
|---|
| Current studya) | Castellan et al.18) b) | Dickinson et al.40) c) |
|---|
| CYP2C9*1/*2 | 0.79 | 0.82 | 0.85 |
| CYP2C9*1/*3 | 0.60 | 0.56 | 0.55 |
| CYP2C9*2/*2 | 0.58 | 0.70 | 0.70 |
| CYP2C9*2/*3 | 0.39 | 0.39 | 0.40 |
| CYP2C9*3/*3 | 0.19 | 0.13 | 0.10 |
a) Each FA2C9 value was estimated by Eqs. 2 and 3. b) Each FA2C9 value was estimated with the data derived from in vivo pharmacokinetic studies. c) Each FA2C9 value was estimated with the data derived from in vitro yeast expressing system.
Table 3 shows the predictions for AUCXM/AUCEM of fluvastatin. These predicted values were compared to the observed values of 3R,5S-fluvastatin that were derived from a clinical study.11) Our predicted values were within a range of 50–200% of the previously determined values (Supplementary Fig. 1).
Table 3. Prediction of Increase in the
AUC of Fluvastatin in Carriers with the CYP2C9 Genetic Variants Compared to That in Patients with the CYP2C9 Wild-Type at the Same Dose (
AUCXM/
AUCEM)
| CYP2C9 genotypes | Predicted AUCXM/AUCEM | Observed AUCXM/AUCEM a) |
|---|
| CYP2C9*1/*2 | 1.16 | 1.15 |
| CYP2C9*1/*3 | 1.35 | 1.34 |
| CYP2C9*2/*2 | 1.37 | 1.04 |
| CYP2C9*2/*3 | 1.65 | 2.24 |
| CYP2C9*3/*3 | 2.06 | 3.08 |
a) Each observed value was calculated based on the AUC values of 3R,5S-fluvastatin reported in an in vivo clinical interaction study.11)
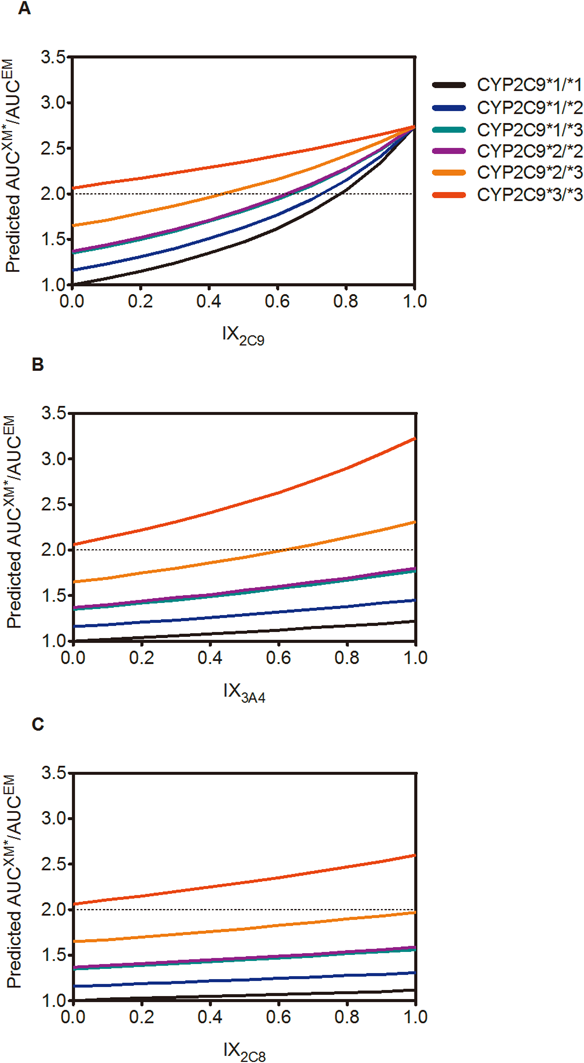
Figure 1 shows the predictions for the AUCXM*/AUCEM of fluvastatin. Although the CR3A4 and CR2C8 were much smaller than the CR2C9 in the carriers with CYP2C9*1/*1, these results did indicate that potent CYP3A4 and CYP2C8 inhibitors might be able to cause 3.23-fold and 2.60-fold increases in the AUC of fluvastatin in the CYP2C9*3/*3 carriers, respectively, compared to the AUCEM.
Even if the highest Cmax,blood was used, the IX2C9 of candesartan was only estimated to be 0.01. Based on these results, we considered the inhibition of CYP2C9 by candesartan in clinical situations to be negligible.
Calculated concentration and the formation rate of 6α-hydroxypaclitaxel in pooled HLMs in the absence of inhibitors were 958±62.1 nM and 31.9±2.1 pmol/mg protein/min, respectively (mean±S.D., n=3). IC50 values of telmisartan and candesartan for paclitaxel 6α-hydroxylation catalyzed by CYP2C8 were estimated to be 46.0 and 176.7 µM, and the Ki values for CYP2C8 were estimated to be 23.0 and 88.4 µM, respectively. The IX2C8 of telmisartan for the various dosages were calculated using Eq. 6, whereas the IX2C8 of candesartan was estimated to be 0.00 (not measurable) even if the highest Cmax,blood was used. Based on these results, we believe the inhibition of CYP2C8 by candesartan in clinical situations to be negligible.
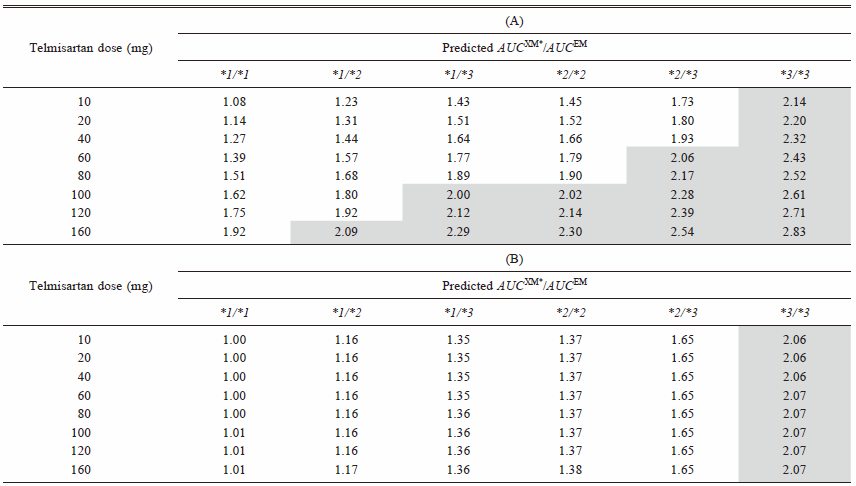
Table 4 shows the predicted AUCXM*/AUCEM of fluvastatin after coadministration of various dosages of telmisartan.
Table 4. Prediction of Increase in the
AUC of Fluvastatin in Carriers with the CYP2C9 Genetic Variants (
AUCXM*/
AUCEM) after Coadministration of Telmisartan
AUCXM*/AUCEM of fluvastatin was predicted by Eq. 5 when coadministered with various dosages of telmisartan by using (A) total (bound and unbound) concentration and (B) unbound concentration. Highlighted cell represents that it exceeds the boundary of the Pharmacokinetic Interaction Significance Classification System (PISCS) defined ‘warning/cautions’ for statins.8)
DISCUSSION
Results of the present study show the importance of the CYP2C9 genetic variants on the fluvastatin pharmacokinetics. We estimated CR2C9 and CR3A4 to fluvastatin metabolism from two different clinical studies,21,22) while CR2C8 from our in vitro experiment using MAB-2C8. Results indicated that MAB-2C8 only inhibited the M-3 formation (data not shown), which was consistent with our previous report that also used MAB-2C8.4) These values are consistent with the values that have been reported for the product labeling (Lescol®) in the U.S.A.38) Assuming that CYP2C9 genotypes only affect the activity of CYP2C9, activities of CYP2C8 and CYP3A4 would conserve among carriers of all CYP2C9 genotypes. CR2C9 in carriers of CYP2C9*2 and CYP2C9*3 variants would be low relative to those with CYP2C9 wild-type, indicating that the metabolic pathways by CYP2C8 and CYP3A4 would be more important than CYP2C9 on fluvastatin metabolism in carriers of CYP2C9 variants. This assumption is consistent with our result (Fig. 1A) and the previous result reported by Kumar et al.,39) where the inhibitory effects of CYP2C9 inhibitors decreased in carriers of CYP2C9*1/*3 and CYP2C9*3/*3 compared to the wild-type carriers.
The FA2C9 of the 6 diplotypes of CYP2C9 that were calculated in this study were in a good agreement with them estimated in meta-analyses with in vitro data derived from yeast expressing system40) and in vivo data18) (Table 2). Although the values obtained in our current study were calculated based on an in vitro experiment with a single lot of rCYP2C9 proteins for each CYP2C9 variant that did not distinguish the concentrations for each of the fluvastatin enantiomers, the AUCXM/AUCEM predicted when using Eq. 4 were within a range of 50–200% of the observed AUCXM/AUCEM of the active enantiomer, 3R,5S-fluvastatin, that was derived from a previous clinical study11) (Table 3). Thus, these results indicate that the predicted AUCXM/AUCEM of fluvastatin in this study correspond to the values of 3R,5S-fluvastatin and that in vitro prediction of the in vivo situation is reasonably good.
Muscle toxicity, which is one of the leading causes of statin discontinuation, has been suggested to be dose-dependent.5) Based on our predictions, the AUC of fluvastatin in the carriers of the CYP2C9*3/*3 variant was estimated to be 2.1-fold higher versus the carriers with the CYP2C9*1/*1 at the same dosage (Table 3). Hisaka et al. initially proposed the Pharmacokinetic Interaction Significance Classification System (PISCS).8) According to this classification system, the combinations of the CYP3A4 inhibitors and the statins that are primarily metabolized by CYP3A4 caused a >7-fold and >2-fold increase of AUC were categorized as ‘contraindication’ and ‘warning/caution,’ respectively. The predicted AUCXM/AUCEM in the carriers with the CYP2C9*3/*3 variant was greater than the boundary for the ‘warning/cautions’ (Table 3), this indicates that further decrease of fluvastatin clearance such as DDI in those patients might increase the risk of ADRs of fluvastatin even though the effect of DDI is negligible in population of CYP2C9*1/*1 careers. In fact, Miroševic Skvrce et al. have found an increased risk of ADRs when carriers of CYP2C9 variant alleles were given CYP2C9 inhibiting medicines concomitantly.12)
Ohno et al.7) and Hisaka et al.8) have reported that careful attention needs to be paid to the co-administration of CYP3A4 inhibitors when using statins that are mainly metabolized by CYP3A4. It has also been reported that fluvastatin should be considered to be a good choice for patients who need to be treated by CYP3A4 inhibitors such as calcineurin inhibitors. Our prediction indicated that co-administration of a potent CYP3A4 inhibitor with an IX3A4 of 1.0 in patients genotyped as CYP2C9*3/*3 would see a 3.2-fold increase in the AUCXM*/AUCEM (Fig. 1B). Although negligible interaction has been reported between fluvastatin and itraconazole in healthy volunteers, in which the information about CYP2C9 genotype was not available, our results do suggest that co-administration of potent CYP3A4 inhibitors would increase the risk of fluvastatin-induced ADRs in homozygous carriers of CYP2C9*3. This finding can be explained by a relative increase in CR3A4, along with decreasing CR2C9, to fluvastatin metabolism in carries of that mutative allele relative to the wild-type carriers, as discussed above. To date, 60 genetic variants for CYP2C9 have been reported, while our current study only used 6 diplotypes. Therefore, a further study will need to be undertaken in order to definitively determine the effect of other CYP2C9 genetic variants on the fluvastatin pharmacokinetics.
To investigate the effect of telmisartan and candesartan on the fluvastatin pharmacokinetics, we calculated the IX2C9 and IX2C8 for both of the drugs. Although candesartan showed inhibitory effects for CYP2C9 and CYP2C8, both of the IXs of candesartan were essentially zero, even when using the highest Cmax,blood after multiple oral administrations for the calculation. Since telmisartan is a substrate of OATP1B329) and accumulates in the liver, the IXs of telmisartan were estimated by using [I]in and [I]in,u with the various dosages. Moreover, due to the nonlinear pharmacokinetics of telmisartan,34) we also used the Cmax,blood after multiple oral administrations with the various dosages of telmisartan for our calculations of the [I]in. As seen in Table 4A, our results using [I]in for the prediction showed that when 160 mg of telmisartan was co-administered with fluvastatin in carriers of any mutated CYP2C9 alleles, the PISCS classified the results as ‘warning/caution’,8) whereas the inhibitory effects were negligible when using [I]in,u for the prediction (Table 4B). A recent case report13) has suggested that a pharmacokinetic interaction occurred between fluvastatin and telmisartan, but not candesartan, in a subject with the CYP2C9*1/*3 genotype who was taking 20 mg of telmisartan. Based on our predictions using the theoretically highest concentration, [I]in, the AUCXM*/AUCEM of fluvastatin in subjects genotyped as with the CYP2C9*1/*3 variants after the co-administration of 20 mg of telmisartan would be 1.51. It was slightly higher than the 1.35 that was predicted in the absence of inhibitors or in the presence of candesartan in subjects with the CYP2C9*1/*3 variants (Table 3). Furthermore, an in vivo interaction study showed only 1.09-fold increase in AUC of warfarin with the co-administration of 120 mg of telmisartan.41) These findings indicate that it is unlikely to occur the interaction between telmisartan and fluvastatin via CYP enzymes in clinical situations. Since some OATPs and an ATP-binding cassette sub-family G member 2 (ABCG2) efflux transporter are associated with disposition of fluvastatin,12,42,43) the interactions via those transporters between fluvastatin and telmisartan might occur in vivo situation. An additional study will need to be undertaken in order to further elucidate the DDI on the fluvastatin pharmacokinetics.
In summary, we found that CYP2C9 genotype affected DDI-potential of fluvastatin. The impact of CYP3A4 inhibitors would be stronger if the total clearance of fluvastatin to a greater extent depended on CYP3A4 rather than CYP2C9 in patients with a poor metabolizer genotype CYP2C9*3/*3.